Texto universitario

_____________________________
Módulo 1. El enlace químico
Átomo
El concepto de la configuración electrónica orbital de un átomo se presenta en muchos libros de texto de química y no es necesario explicarlo aquí en detalle. Sin embargo, hay dos constituyentes principales de los átomos: orbitales en los que se alojan los electrones y un núcleo en el que se alojan especies como los neutrones y los protones. La química nuclear se ocupa del comportamiento de los neutrones y los protones y, a veces, del comportamiento y la estructura de los electrones. Los electrones están dispuestos fuera del núcleo atómico en varios orbitales. Cada orbital o nube, descrito por cuatro números cuánticos, tiene energía definida y puede acomodar dos electrones con un espín antiparalelo. La transición de un electrón de un orbital al otro resulta en la emisión o absorción de una cantidad definida de energía. La magnitud de esta energía corresponde a la diferencia entre la energía de los dos orbitales en cuestión. Esta energía se emite en forma de radiación electromagnética. Los átomos se combinan para formar un compuesto, mediante la transferencia de electrones de un átomo a otro seguido de una atracción electrostática (electrovalencia) o compartiendo electrones (covalencia). Durante las reacciones químicas, los electrones presentes en los orbitales más externos se reorganizan.
A) Estructura nuclear
Para establecer completamente la constitución de un núcleo atómico, es necesario conocer la naturaleza de los constituyentes, las fuerzas que los unen y las leyes que gobiernan su comportamiento. Los constituyentes nucleares fundamentales son los protones y los neutrones, y las leyes que gobiernan sus interacciones son las de la mecánica cuántica. Sin embargo, la naturaleza precisa de las fuerzas nucleares aún no se comprende completamente. Gran parte de la información conocida está incorporada en varios modelos nucleares; cada uno de estos tiene ventajas, pero ninguno de ellos puede explicar todos los datos experimentales disponibles de un núcleo.
B) Modelo de capa
Entre los diversos modelos nucleares, el modelo de capa es más popular porque puede explicar la mayor parte del comportamiento nuclear del átomo y ha ayudado a sintetizar nuevos isótopos, cuya existencia se desconocía. La base de este modelo sigue conceptos casi similares a la disposición de los electrones en un átomo. Por lo tanto, puede ser más fácil explicar el modelo de capa comparándolo con una estructura electrónica atómica. Por lo tanto, quizás sería mejor revivir brevemente el conocimiento básico de una estructura atómica.
El átomo consta de electrones y giran alrededor del núcleo con diferentes orbitales dependiendo de la cantidad de electrones presentes en el átomo. Los protones y los neutrones están presentes en el núcleo. Los diversos orbitales designados para electrones son s, que puede albergar un máximo de 2 electrones, p, que puede albergar un máximo de 6 electrones, d, que puede albergar un máximo de 10 electrones, y f orbital que puede albergar 14 electrones como capacidad máxima. Los átomos con un número total de electrones, es decir, 2 (helio), 10 (neón), 18 (argón), 38 (criptón), 54 (xenón) y 86 (radón) son los elementos más inertes y estables, y otros elementos que contienen menos o más cantidad de electrones suelen ser menos estables. Estos números también se conocen como números mágicos. Al igual que la disposición de los electrones en un átomo, el modelo de capa también supone la revolución del protón o el neutrón en algún orbital específico, de modo que no más de 2 nucleones (es decir, protón o neutrón) estarían presentes en un orbital. A medida que se agregan nucleones al núcleo, primero ocupan la energía más baja permitida por el Principio de exclusión de Pauli. Esto significa que cada nucleón posee un número cuántico único que representa su movimiento. Cuando el orbital de la capa está completamente lleno, el núcleo alcanza una gran estabilidad. Se supone que, al igual que los electrones, los nucleones poseen espín (1/2) y poseen un número cuántico como l y m. También se supone que dentro del núcleo existen orbitales separados para protones y neutrones. Por lo tanto, los primeros dos protones se llenarán en el nivel cero (es decir, 0, 0, 0, +1 2 y −1 2 = 2 protones). Asimismo, los siguientes seis protones se llenarán en el nivel uno. De esta manera, los protones pueden llenarse en todos los demás niveles posibles. Teniendo en cuenta la disposición de seis capas, el número de nucleones distribuidos seguiría como se muestra aquí:
∗ nivel 0: 2 estados (l = 0) = 2
∗ nivel 1: 6 estados (l = 1) = 6
∗ nivel 2: 2 estados (l = 0) + 10 estados (l = 2) = 12
∗ nivel 3: 6 estados (l = 1) + 14 estados (l = 3) = 20
∗ nivel 4: 2 estados (l = 0) + 10 estados (l = 2) + 18 estados (l = 4) = 30
∗ nivel 5: 6 estados (l = 1) + 14 estados (l = 3) + 22 estados (l = 5) = 42
Si los nucleones se llenan de esta manera para todas las capas posibles, se observa que algunas capas contienen un número específico de nucleones como se muestra aquí:
2
8 = 2 + 6
20 = 2 + 6 + 12
28 = 2 + 6 + 12 + 8
50 = 2 + 6 + 12 + 8 + 22
82 = 2 + 6 + 12 + 8 + 22 + 32
126 = 2 + 6 + 12 + 8 + 22 + 32 + 44
184 = 2 + 6 + 12 + 8 + 22 + 32 + 44 + 58
Si los protones y los neutrones se llenan de esta manera, se observa que aquellos núcleos que tienen capas completamente ocupadas tendrán el número de nucleones como 2, 8, 20, 28, 50, 82, 126 y 184. Se observa que los núcleos que contienen este número específico de nucleones son altamente estables en comparación con aquellos que tienen un nucleón menos o uno más. Este número es reconocido como Número Mágico. El modelo de capa predice así la posibilidad de nuevos isótopos que aún no han sido descubiertos como los isótopos de los números mágicos 184 y 126. Se observa que los núcleos que contienen números pares de protones y neutrones son más estables que los que tienen números impares, lo que se debe al efecto de emparejamiento. Además, los núcleos que tienen números de protones y neutrones iguales a uno de los números mágicos se denominan doblemente mágicos y son muy estables.
Energía de enlace del núcleo
Se observa que la masa de un átomo es menor que las masas combinadas de los constituyentes separados del átomo. Esta diferencia de masa se denomina “defecto de masa” y puede expresarse en términos de “energía” utilizando la conocida relación masa-energía de Einstein, es decir,
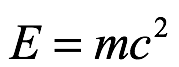
donde “c” es la velocidad de la luz y “m” la masa. Esta energía es responsable de unir los nucleones en el núcleo y se llama "energía de enlace".
C) Nube electrónica
Nuestro enfoque ahora se centra en las propiedades enormemente importantes de la nube de electrones que rodean un núcleo. Necesitamos hacer más precisa la naturaleza y la estructura de esas nubes, porque no son solo regiones de niebla arremolinada. Los electrones rodean el núcleo en capas, más bien como nubes que se encuentran una encima de otra, pero que rodean todo el átomo. El conceptos de que un electrón es una “nube” necesita una rápida explicación. La nube es realmente una nube de probabilidad: donde es densa, es probable que se encuentre el electrón; donde es escasa es poco probable que se encuentre el electrón.
Las leyes de la mecánica cuántica ordenan que hasta dos electrones rodean el núcleo en la capa más baja, hasta otros ocho en la siguiente capa circundante, y luego otros 18 en la siguiente capa. No necesitaremos ir más allá de eso, pero es un patrón similar con variaciones continúa indefinidamente a medida que crece el número de electrones. Este patrón significa que en el hidrógeno un solo electrón rodea el núcleo. En el carbono, con sus seis electrones, dos electrones formando la nube en el nivel más bajo y cuatro más forman las nubes circundantes en la capa exterior. Se podría pensar en los átomos como núcleos de nubes rodeados de capas de nubes en forma de cebolla, cada capa interna se completa antes de que comience la siguiente capa. El por qué existen estos números característicos (2,8,18…) para capas sucesivas no tiene por qué preocuparnos, sino que se entiende plenamente en términos de mecánica cuántica.
Ahora todo debe estar claro: el diseño de la tabla periódica representa el llenado de las capas de las nubes, con un electrón presente en la capa a la izquierda de la tabla y capas completas a la derecha. Por razones técnicas que se entienden completamente, pero es una distracción aquí, el orden en que se completan las capas de nubes se confunde un poco después de las dos primeras filas de la tabla, y aunque las longitudes de las filas son los números que ya hemos visto, a saber, 2,8,18…, y se pueden discernir, se encuentran en un orden comprendido en un patrón periódico.
El punto crucial es que los elementos que se encuentra uno debajo del otro tienen patrones muy similares de cobertura de nubes. Ese es el origen de las relaciones familiares: el oxígeno y su primo azufre en la fila de abajo tienen el mismo patrón de nubes, es solo que los últimos seis electrones del azufre se encuentran en un nivel más alto que los seis finales del oxigeno. Del mismo modo, los últimos cinco electrones del fósforo se encuentran en una capa de nivel más bajo que los últimos cinco de nitrógeno en la fila sobre él.
A menudo se dice que los átomos son en su mayoría espacio vacío. Eso simplemente no es cierto. Las distribuciones de electrones en forma de nube llena todo el espacio alrededor del pequeño núcleo del tamaño de una mosca en un estadio de futbol. Es cierto que las nubes son muy delgadas en partes; pero está ahí y omnipresente. La afirmación de que un átomo es en su mayoría espacio vacío surge de la visión anticuada de que los electrones son como pequeños planetas puntiagudos que zumban alrededor del núcleo a grandes distancias de él, con mucho espacio vacío en medio. La mecánica cuántica sustituye esta figura por distribuciones de probabilidad en forma de nube que hemos descrito, nubes que, aunque muy atenuadas en partes, llenan todo el espacio.
Cómo los átomos forman enlaces
La principal preocupación de la química no es tanto con átomos individuales, sino con los compuestos que forman al entrar en una variedad de enlaces entre sí. Hay literalmente millones de enlaces que han sido identificados en compuesto y muchos más que sabemos que existen pero no han sido identificados y nombrados. La riqueza de nuestro entorno se debe a esta enorme colección de compuestos, y los químicos pasan la mayor parte de sus horas construyendo nuevas combinaciones de átomos o derivando compuestos para ver cómo se construyen. Para hacer esto de manera efectiva, necesitan comprender cómo los átomos se unen entre sí y qué controla los enlaces químicos, que pueden formar entre sí.
¿Qué mantiene unidos a los átomos para formar compuestos identificables? ¿Puede haber alguna combinación de átomos, o hay razones para la moderación de la naturaleza que los químicos a pesar de toda sus intromisión no pueden eludir? ¿Por qué hay variedades en el mundo de las sustancias, pero aparentemente no verdades aleatorias? Estas preguntas se pueden inferir: ¿por qué no todos los átomos del universo simplemente se agrupan en una enorme mesa sólida?
“Llamamos enlace químico a la fuerza por la que los átomos de un compuesto se mantienen unidos. Se trata de fuerzas electromagnéticas que dan lugar a diferentes tipos de enlaces químicos. Los principales tipos de enlaces químicos entre átomos son tres: enlaces iónicos, covalentes y metálicos. Se trata de enlaces fuertes y duraderos, que unen a un átomo con otro átomo o grupo de átomos. El tipo de enlace que se genere influirá fuertemente en las propiedades de los compuestos químicos formados. Los átomos tienden a formar enlaces químicos porque cuando se unen a otros átomos alcanzan una situación más estable, es decir, la que les supone un consumo de energía menor. Esto ocurre cuando el número de electrones de su último nivel es igual a ocho, lo que se conoce como la regla del octeto. Es cierto que no es una norma que se pueda aplicar a todos los átomos, pero sí a la mayoría. Para cumplir con la regla del octeto para enlaces, los átomos pueden ceder electrones, ganarlos o incluso compartirlos con otro átomo. Los enlaces iónicos ocurren cuando un átomo gana o pierde electrones. Como resultado de esta transferencia de electrones, se forman iones o, lo que es lo mismo, partículas cargadas. Otro modo que tienen los átomos para formar enlaces químicos es compartiendo electrones. De esta manera, crean enlaces covalentes. Los electrones no se quedan inmóviles en un punto, sino que se mueven entre los átomos. Esto da lugar a los principales tipos de enlaces covalentes: el enlace covalente polar y el apolar. La estructura del enlace metálico se debe a que los átomos de los metales tienen pocos electrones en la última capa, que pierden fácilmente. Como resultado, se convierten en cationes que se distribuyen por el espacio formando una especie de red, mientras que los electrones que han perdido crean a su alrededor una nube de electrones que puede moverse por toda la red. Así los cationes, con carga positiva, queden unidos mediante la nube de electrones, con carga negativa, que los envuelve[1]”.
El resumen anterior esconde mucho conocimiento, las respuestas a estas preguntas están en un tipo de discurso que discute en esas capas de nubes. En términos generales, hay ventajas energéticas en un átomo que adquiere una capa de la nube completa. Puede hacerlo de varias maneras. Una es arrojar electrones de la capa más externa. Esto es probable que suceda si no hay muchos electrones en esa capa para empezar, lo que significa que es más probable que suceda con átomos de elementos a la izquierda de la Tabla Periódica, el comienzo de cada nueva fila y cada nueva capa de nube. Alternativamente, si ya tiene muchos electrones en su capa más externa, entonces podría ganar electrones de algún lugar y así completar su capa. Es probable que eso suceda si la capa está casi llena, que es el caso de los átomos de los elementos más a la derecha de la Tabla Periódica hacia el extremo derecho de una fila. Hay forma de completar sus capas: los átomos podrían competir por electrones de la capa externa de otro. Eso podría suceder cuando un átomo es reacio a liberar un electrón por completo porque no hay ninguna ventaja de energía en él. Esa sutil propiedad el carbono forma la mayoría de sus extraordinarios enlaces de esta manera.
Como hemos visto, los átomos son eléctricamente neutros, con la carga negativa total de todos sus electrones igualando y cancelando la carga positiva total de todos los protones en el núcleo. Cuando un átomo gana o pierde un electrón, el equilibrio de cargas se altera y el átomo se convierte en un ion. Un ion es implemente un átomo cargado eléctricamente; se llama así porque se moverá en respuesta a un campo eléctrico, “ion” es la palabra griega para referir al “moverse”. Un átomo que ha ganado uno o más electrones está cargado negativamente y se llama anión. Uno que ha perdido uno o más electrones está cargado positivamente y se llama catión. Se puede resumir que es probable que los elementos a la izquierda de la Tabla Periódica pierden su pocos electrones más externos y, por lo tanto, se convierten en cationes; aquellos a la derecha de la Tabla Periódica con nubes más externas casi completas es probable que ganen electrones y se conviertan en aniones.
Nos hemos encontrado con uno de los grandes mecanismos de enlace: debido a que las cargas opuestas se atraen entre sí, y los cationes y aniones están cargados de manera opuesta, se deduce que los átomos que forman estos iones se agrupan en un compuesto. La sal común, cloruro de sodio, es un excelente ejemplo de este tipo de formación de compuestos. El sodio (Na, de su nombre latino natrium) se encuentra a la izquierda de la Tabla Periódica, y libera fácilmente su único electrón más externo para formar un catión de sodio, denotado Na+. El cloro (Cl) se encuentra a la derecha de la Tabla Periódica, y felizmente acomoda un electrón para completar su capa externa y, por lo tanto, convertirse en un anión cloruro, Cl-. Nótese el pequeño cambio de nombre de cloro a cloruro). Los iones se agrupan y forman cloruro de sodio, una tabla rígida de iones unidos por su atracción mutua. Ya hemos enfatizado que los átomos son muy pequeños, y que incluso pequeñas muestras de una sustancia contienen muchos de ellos. Somos un Atlas entre las estrellas cuando se trata de iones, por que cuando recogemos un grano de sal, estamos sosteniendo más iones que estrellas en el universo visible.
[1] https://www.zschimmer-schwarz.es/como-se-forma-la-materia-tipos-de-enlaces-quimicos-ejemplos-y-caracteristicas/
1.1 La química
La codicia inspiró a la humanidad a embarcarse en un viaje extraordinario que afecta a todos hoy en día. La particular variedad de codicia a la que referimos, fue la búsqueda de inmortalidad y el logro de riquezas ilimitadas. La supuesta ruta de ambas empresas fue el intento de manipular la materia para proporcionar elixires al cuerpo y convertir plomo en oro de manera rentable. Nunca se logró ningún objetivo, pero el incesante trabajo hizo surgir una verdadera ciencia: la química. Siendo el principal instrumento de la transición de la alquimia a la química el concepto de equilibrio. La capacidad de sopesar las cosas con precisión puso en las manos de la humanidad el potencial de unir números, geometría y álgebra a la materia. La importancia de ese logro no debe ir sin ser comentado, ya que de hecho, es de lo más significativo unir números al aire, al agua, al oro y a cualquier tipo de materia en forma de datos. Así, a través de la vinculación de números y materia las matemáticas y la física cuantitativamente experimentaron rigurosamente para crear teorías que iluminaran los datos.
Pensar la materia después de haber sufrido una transformación de una sustancia en otra, llevó al concepto principal que subyace a todas las explicaciones en la química: el átomo. El concepto de átomo había flotado sin fundamento desde los griegos, que lo consideraron por error la parte indivisible de la materia. John Dalton (1766-1844), a través del análisis de pesos de las sustancias, antes y después de la reacción llegó a la conclusión de que los elementos, los bloques que fundan las sustancias como una conveniencia casi común a todas las explicaciones químicas[1]. Los átomos como elementos o combinados, llamados moléculas, constituyen la materia que podemos ver y tocar. Tan pequeños como son, es bastante erróneo decir elementales de la materia, están compuestos de átomos y esa pista podría mantener que son invisibles a simple vista. Mire las cosas a su alrededor, está usted viendo átomos en lo que llama mesa, una página o pantalla de ordenador. Por su puesto un átomo individual es muy pequeño para verlo, pero la materia se construye a partir de muchos de ellos.
Hay más de 100 tipo de átomos. Sus diferentes estructuras internas es lo que los hacen distintos en su tipo. Cada tipo diferente de átomo corresponde a un elemento diferente. La idea clave de la química es que cuando una sustancia cambia a otra, los átomos en sí mismos no cambian, simplemente intercambian socios o entran en nuevos arreglos. La química tiene que ver con asociación y ruptura de asociación de átomos. Significa que para comprender la química hay que importar conceptos de la física, dado que se basa en el comportamiento electromagnético y sus componentes subatómicos[2]. La importancia crucial de la física, da cuenta de las propiedades del mundo microscópico de átomos y moléculas individuales, dado por la mecánica cuántica. Aunque gran parte de la química se desarrolló en el siglo XIX, hubo poca comprensión de porqué algunas cosas ocurrieron y otras no en los experimentos. Newton y su mecánica tuvo tanto éxito en las órbitas de planetas y estudio de cuerpos en movimiento, que se pensó en aplicarse a la química. A principios del siglo XX los conceptos de Newton aplicados al átomo se desmoronan cuando al extrapolar el sistema solar a la estructura del átomo, los datos fueron contundentes sobre un mundo on una lógica totalmente distinta.
Alrededor de 1927, nació una nueva mecánica que ha demostrado ser enormemente exitosa para explicar cómo los átomos y las partículas subatómicas se estructuran e interactúan. Hasta el día de hoy, la teoría de la mecánica cuántica, no ha sido remplazada debido a su potencial productivo y precisión numérica.
La otra importante y crucial aportación de la física como cimientos de la química, es que permite calcular en el mundo microscópico efectos importantes fuera de la escala del hombre con la llamada termodinámica. La termodinámica es la ciencia de la energía y las transformaciones que puede sufrir. Mientras la química hace sus cimentos en la física para sus explicaciones, llega sobre ella una en nueva ciencia, la biología que sin la química no podría procesar sus explicaciones[3]. Los organismos se construyen a partir de átomos y moléculas, y con esas estructuras funcionan en virtud de la lógica química subyacente.
Para nuestros propósitos, y para comprender la estructura general de la química en aras de ganar profundidad, es útil apreciar sus divisiones en varias de sus ramas, en términos generales en función de sus preocupaciones de estudio. Aunque las fronteras intelectuales y ciencias de estas ramas, ahora mismo son casi inexistentes.
La química física se encuentra en la interfaz de la física y la química. Se ocupa de la química y en gran medida tiene que ver con la mecánica cuántica para explicar las estructuras de átomos, moléculas y su termodinámica para evaluar el papel y el despliegue de la energía. También se refiere a las velocidades a las que tienen lugar las reacciones, tanto a nivel microscópico como macroscópico. En este nivel más bajo, en el microscópico se reconstruye la vida íntima de las moléculas en sus reacciones y equilibrios. Una actividad importante de la química física es su contribución a la interpretación de las técnicas de investigación, en particular la espectroscopia. Tal es la sofisticación actual de estas técnicas que los químicos físicos desarrollan todo un arsenal de técnicas analíticas para llevar a cabo interpretaciones de los datos y construir predictores en apoyo al diseño de sustancias sintéticas[4].
Química orgánica, es la parte de la química que se refiere a los compuestos de carbono. Ha sido definida por un elemento que puede comandar toda una división de conocimiento, es el resultado de su carácter suave y complejo capaz de formar cadenas y anillos de sorprendente estructuración. Y esta complejidad es lo que permite a los organismos ser considerados vivos, y por lo tanto los compuesto de carbono son la infraestructura estructural y reactiva de la vida. Tan extensos son los compuestos de carbono, que actualmente se numeran en millones, que no es de extrañar que toda una rama de la química ha desarrollado técnicas especiales, sistemas de nomenclaturas y actitudes científicas para su estudio. Tal es la complejidad de las moléculas a las que constituye el carbono, que originalmente se pensaba que solo la naturaleza podía formarlas. Es decir, según este punto de vista, son productos de los organismos. El principio del vitalismo introducido en 1828, demostró que un mineral simple podía transformarse en un compuesto “orgánico” característico (es decir, urea). Aunque la diputada se extendió durante algún tiempo, desde entonces la “orgánica” de la química ha sido un arcaísmo; pero los arcaismos convenientes son difíciles de desalojar y el término sobrevive, pero ahora no significa nada más que “un compuesto de carbono[5]”.
Eso deja el resto de los elementos, los más de cien elementos que no sean carbono. Su estudio es la química inorgánica. Como se podría sospechar acerca de una rama de estudio de más de 100 elementos muy diferentes, la química inorgánica es un campo dinámico y extenso. Una división más es la química de estado sólido, estudia los materiales superconductores, conductores y semiconductores de la industria electrónica. En la frontera entre las divisiones, sin embargo, hay compuestos que son intrincados de átomos de carbono pero que incluyen átomos de varios metales, son catalizadores esenciales en la industria química; algunos son cruciales para el funcionamiento de los agonismos[6]. Aquí se encuentra el campo interdivisional de la química organometálica, que en su momento representa una colaboración altamente fructífera entre químicos orgánicos e inorgánicos. Esta lista para nada agota las principales divisiones de la química, pero si pueden agrupar las principales ramas.
La química analítica es el descendiente moderno de la antigua búsqueda de averiguar lo que hay. ¿Qué está presente en un mineral? ¿Podría haber plata o hafnio? ¿Qué está presente en el petróleo crudo? ¿Qué es ese compuesto, puede reducir los arreglos de átomos? Estas son algunas preguntas que los químicos analíticos podrían tratar de responder. Aunque los tubos de ensayo, los matraces y las réplicas todavía figuran en sus enfoques, muchas de sus investigaciones se llevan a cabo ahora en máquinas sofisticadas, algunas de la cuales utilizan espectroscopia y otras técnicas desarrolladas por químicos inorgánicos y físicos. La química analítica se deriva de la química forense, en la que las técnicas de química analítica se utilizan con fines legales, para rastrear o exonerar a los sospechosos y para analizar las escenas de crímenes[7].
La bioquímica es la heredera de la química orgánica a la biología, a veces con una pizca de química inorgánica. Se refiere totalmente a las estructuras y reacciones que constituyen seres vivos, resolviendo las vías metabólicas que convierten los alimentos en acciones, incluyendo las acciones confinadas en el cerebro para producir el pensamiento. Los organismos siguen siendo un reservorio muy importante de moléculas orgánicas, ya que la naturaleza ha tenido de millones de años para explorar nichos estructúrales y, los bioquímicos juegan el papel central tanto en el descubrimiento de lo que hay allí, como en el trabajo de cómo se hizo, por ejemplo el control de las abejas obreras del cuerpo, es decir, las proteínas que llamamos enzimas. Una preocupación antropocéntrica pero importante sobre la extinción de especies es que elimina las fuentes de moléculas intrincadas que han tardado millones de años en surgir[8].
La tabla periódica retrata una característica extraordinaria de la materia, que los elementos están relacionados entre sí. Ahora estamos tan familiarizados con la tabla que esa característica se olvida fácilmente. Sus apariencias muy diferentes son diferencias superficiales, ya que cuando se investigan las reacciones en las que participan y las moléculas que forman, resulta que hay profundas similitudes entre estos parientes. Esas similitudes provienen de las estructuras de sus átomos, y para entenderlas es a estos átomos a los que ahora debemos mirar.
La estructura básica de un átomo consiste en un núcleo rodeado por una nube de electrones. Este es el modelo de un átomo identificado por primera vez por Ernest Rutherford en 1911. El núcleo está cargado positivamente, los electrones se cargan negativamente y es la atracción entre estas cargas opuestas la responsable de la existencia y supervivencia del átomo[9]. Como es bien sabido, los átomos son muy pequeños, hay más de un millón de átomos de carbono en el punto de tinta de la versión impresa de esta frase. Un núcleo es aún más pequeño, la metáfora clásica dice que si un átomo se agrandará al tamaño de un estadio de fútbol, el núcleo sería del tamaño de una mosca en el centro del campo.
Un núcleo consta de dos tipos de partículas subatómicas: protones y neutrones. Como sugieren los p y n en su nombres, los protones son de carga positiva y los neutrones de carga neutra. Los neutrones tiene la capacidad de transformarse en protones, pero esto está lejos de nuestro manuscrito. Aparte de eso, son muy similares, con casi la misma masa. Están firmemente unidos en el núcleo, y requieren un gran esfuerzo, algo así como una explosión nuclear para liberarlos. En la mayor parte de la química, con sus liberaciones relativamente débiles de energía, el núcleo permanece inalterado y es un participante pasivo pero importante en los procesos que se están llevando a su alrededor en tubos de ensayo y matraces.
El número de protones en el núcleo determina la identidad química del átomo. Por lo tanto, un átomo de hidrógeno tiene un protón, un átomo de helio tiene dos, un átomo de carbono tiene seis, nitrógeno siete, oxígeno ocho, y así sucesivamente hasta el elemento 118. El número de protones en el núcleo se denomina número atómico del elemento. A la vez, llegamos a la primera característica extraordinaria de los elementos: se pueden ordenar de acuerdo a su número atómico. Es decir, hay un orden interno de la materia y no algo aleatorio. Se encuentran en una secuencia definida, y el número atómico permite a los científicos identificar la marca del elemento. Los neutrones son solo pasajeros en esta estructura electrónica, pero al ser casi el mismo número de neutrones que los protones en el núcleo, si bien no afecta su número al número atómico, su masa sí participa. A las diferencias de versiones de los átomos por sus neutrinos, se les denomina isótopos del mismo elemento, porque (del latín “esos” que significa igual + topos “lugar”) en el mismo lugar de la tabla.
El número atómico, el número de protones y por lo tanto la carga positiva del núcleo, determina el número de electrones que lo rodean. Un electrón tiene la misma carga eléctrica que un protón, pero de opuesto signo. Por lo tanto, para que un átomo sea eléctricamente neutro, el número de electrones fuera del núcleo debe ser el mismo que el número de protones dentro del núcleo. Es decir, el número de electrones es igual al número de protones dentro del núcleo. Los electrones son mucho más ligeros que los protones y neutrones (por un factor casi 2,000), por lo que su presencia apenas afecta a la masa de un átomo. Tienen un profundo efecto en las propiedades químicas y físicas del elemento, casi toda la química se puede rastrear a su comportamiento[10].
Como hemos mencionado, todas las reacciones químicas dejan los núcleos intactos. En otras palabras, no cambian las identidades de los elementos, no así los casos de transmutación, resultado del cambio de la energía nuclear y física de neutrones. Los químicos tienen un papel vital qué desempeñar para hacer frente a las consecuencias de los procesos nucleares, especialmente en la preparación de combustible para centrales eléctricas nucleares.
Ahora enfoque su atención en la nube de probabilidad de electrones que rodean un núcleo. Necesita precisar la naturaleza de la nube y la estructura de esas nubes, ya que de ellos depende el papel de las capas, las nubes reales se encuentran una sobre la otra, pero rodeando todo el átomo. El concepto de electrón siendo una nube necesita aterrizar en una nube de probabilidad; donde define la predicción de encontrar un electrón[11]. Las leyes de la mecánica cuántica (su predictor) ordenan que el número máximo de electrones que rodean al núcleo en la capa más baja (capa electrónica K) es de dos, hasta ocho más en la siguiente capa circundante (capa L), luego hasta 18 en la siguiente capa (capa M), la capa N con 32, la capa O tiene 50, la capa P tiene 72 electrones…según el principio de Pauli es igual a 2n2 por capa, donde el número de capa es n. No necesitamos ir más allá de eso, se sigue un patrón similar con variaciones continuas indefinidamente a medida que crece el número de electrones. Este patrón significa que el hidrógeno, por ejemplo, tiene un solo electrón que rodea su núcleo. El carbono, con sus seis electrones, dos electrones forman la nube más baja y cuatro la nube superior. Se podría pensar en un átomos como núcleos rodeados de capas de nubes similares a la cebolla, cada capa interna se completa antes de que comience la siguiente capa. ¿Por qué se dan estos números característicos 2,8,18…, para las capas sucesivas no es necesario preocuparnos, pero se entiende completamente en términos de mecánica cuántica, como el hecho curioso de que hasta el elemento 123 se calcula que estos elementos sintéticos volverán a ser estables.
El diseño de la tabla periódica representa el rellenado de las capas de nubes, con un electrón presente en la capa de la izquierda de la tabla y capas completas a la derecha. Por razones técnicas que se entienden completamente, pero que serían una distracción aquí, el orden en que se completan las capas de nubes se confunde un poco después de las dos primeras filas de la tabla, y aunque las longitudes de las filas son números que ya hemos visto, a saber, 2,8,18…, y se pueden discernir, se encuentran en un orden periódico[12]. El punto crucial es que los elementos que se encuentran uno debajo del otro tienen patrones muy similares de cobertura de nubes electrónicas. Ese es el origen de las reacciones familiares: oxígeno y su primo azufre en la fila de abajo tienen el mismo patrón de nubes, es solo que los últimos seis electrones del azufre se encuentran en un nivel más alto que los últimos seis de oxígeno. Del mismo modo, los últimos cinco electrones del fósforo se encuentran en capas de nivel superior que los cinco últimos del nitrógeno en la fila por encima de él.
A menudo se dice que los átomos son en su mayoría espacio vacío. Eso simplemente no es cierto. Las distribuciones nubladas de electrones llenan todo el espacio alrededor del diminuto núcleo. Es cierto que la nube es muy delgada en partes; pero está allí y todo generalizado. La afirmación de que un átomo es en su mayor parte espacio vacío brota de la visión anticuada del átomo como un pequeño sistema solar. La mecánica cuántica reemplaza esa figura por distribuciones de probabilidad, nubes que, aunque muy atenuadas en partes, llenan todo el espacio.
La principal preocupación de la química no es tanto con los átomos individuales, sino con los compuestos que forman al entrar en una actividad de enlaces entre sí. Hay literalmente millones de esos enlaces que han sido identificados y muchos más que sabemos que existen pero que no han sido identificados y nombrados. La riqueza de nuestro entorno se debe a esta enorme colección de compuesto, y los químicos pasan la mayor parte de sus vidas construyendo nuevas combinaciones de átomos o compuestos de desgarro para ver qué pueden formar entre sí.
Se celebra el 100 aniversario de los documentos de Lewis y Kossel sobre el enlace químico y su influencia en el desarrollo de teorías químicas durante el último siglo[13]. Resultados espectroscópicos y estructurales, que proporcionan información detallada acerca de las estructuras de las moléculas y la distribución de la densidad de electrones en las moléculas, han proporcionado pruebas cada vez más rigurosas de sus modelos de enlaces. Sus ideas esenciales, que fueron formuladas en un marco clásico Newtoniano, han soportado muchas pruebas y resultó ser suficientemente flexible para incorporar las más recientes ideas de la mecánica cuántica. Lo más importante es que proporciona una notación gráfica y un lenguaje para los químicos experimentales, lo que les permitió discutir de manera constructiva y predecir las estructuras de las moléculas y representar gráficamente el curso de las reacciones químicas. Aunque las descripciones de Lewis y Kossel del enlace químico son la piedra angular del estudio de la química, lograron esta distinción envolviendo e incorporando un modo más flexible de ver los enlaces químicos y el desarrollo de una notación universalmente aceptada —en modestas palabras de Newton—, el progreso en la ciencia se consigue estando “sobre los hombros de los demás”.
http://www.libertadacademica.com/EbookLetras11/elements/TablaContenido.html
 13.25.06.png)
1.1 Desarrollo histórico del modelo Lewis/Kossel
1.1.1 La tabla periódica
La edad Victoriana se caracterizó por una obsesión con la clasificación del mundo natural, y animales, rocas y de hecho todo fue recolectado, clasificado y puesto en exhibición en los museos. El estudio de los minerales y el reino animal había comenzado a producir grandes ideas que habían empezado a socavar el punto de vista bíblico tradicional de los orígenes y la edad de la tierra. Por 1863, 56 elementos químicos se habían aislado y caracterizado como únicos sobre la base de sus pesos atómicos y valencias —un número suficiente para desarrollar un sistema de clasificación—. En 1864, John Newlands observó que recurrentes similitudes en sus propiedades químicas podrían resaltarse, que permitiría que los elementos se ordenan de acuerdo con sus pesos atómicos relativos[14]. Un patrón repetitivo se produce para grupos de ocho elementos, de una manera que era una reminiscencia de octetos musicales y por lo tanto descrito por él como Ley de Octavas. En las brechas de estas octavas sugirieron que otros elementos podrían ser descubiertos en el futuro, pero carecían de la autoconfianza para hacer predicciones firmes. Lothar Meyer mostró la misma inseguridad cuando en 1864 no pudo predecir ningún nuevo elemento, cuando desarrolló su tabla periódica en función de las valencias de 28 elementos[15]. Sin darse cuenta del trabajo anterior de Newlands y Meyer; Mendeleev comenzó a clasificar los elementos según sus propiedades químicas mientras escribía los dos volúmenes del libro de texto Principios de Química (1868 a 1870). En una etapa inicial, reconoció las siguientes relaciones basadas en los pesos atómicos de los elementos que tenían propiedades químicas similares[16]:
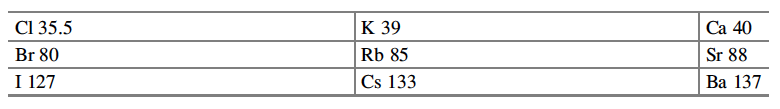
A continuación, se desarrolló una versión extendida de la tabla periódica mediante la incorporación de elementos adicionales que siguieron un patrón similar. Mendeleev hizo una presentación formal de la dependencia entre las propiedades de los pesos atómicos de los elementos, a la Sociedad Química de Rusia el 6 de marzo de 1869. La tabla resultante clasificó los elementos sobre la base de su peso atómico y valencia. Mendeleev dio el paso importante de la predicción de varios elementos nuevos en los huecos que estaban presentes en su tabla, y subrayó su utilidad mediante la predicción específica de propiedades físicas y químicas para estos elementos. Sus predicciones se basan en interpolaciones entre las propiedades físicas y químicas establecidas de elementos, que pertenecían a la misma columna en su tabla. Meses después, Meyer publicó una tabla virtualmente idéntica. Meyer y Mendeleev eran por lo tanto codescubridores de la tabla periódica, pero la decisión de Mendeleev de predecir con exactitud las propiedades del ekasilicio (germanio), ekaaluminium (galio) y ekaboron (escandio) le dio el lugar a él, siendo considerado como el contribuidor más importante por la comunidad química. El premio del Premio Nobel en 1904 a Sir William Ramsay ayudó a consolidar su posición de liderazgo para las generaciones futuras[17]. Estableció que los elementos, si se ordenan de acuerdo a su peso atómico, exhiben una periodicidad aparente de propiedades y sus conclusiones se resumen de la siguiente manera:
1. Los elementos que son similares en cuanto a sus propiedades químicas, tienen pesos atómicos que o bien son de casi el mismo valor (por ejemplo, Pt, Ir, Os) o que aumentan regularmente (por ejemplo, K, Rb, Cs).
2. La disposición de los elementos en grupos de elementos de acuerdo con sus pesos atómicos (con algunas excepciones) pone de relieve las valencias comunes y sus propiedades químicas distintivas. Los elementos más ligeros de estos grupos son Li, Be, B, C, N, O y F.
3. Los elementos que son los más extensamente difundidos tienen pequeños pesos atómicos.
4. El peso atómico de un elemento puede a veces ser modificado por el conocimiento de estos, si son elementos contiguos. Así, el peso atómico del telurio desde estar entre 123 y 126 y no puede ser 128. (La masa atómica del telurio es 127.6, y Mendeleev estaba equivocado en su suposición de que la masa atómica debe incrementar con posición dentro de un periodo.)
5. Ciertas propiedades características de los elementos se pueden predecir a partir de su posición en la tabla periódica.
6. Estaba perplejo acerca de dónde poner los lantánidos conocidos y predijo la existencia de otra fila de la tabla para ellos y los actínidos.
Mendeleev basó las regularidades en la tabla principalmente en los pesos atómicos de los elementos en lugar de sus valencias, porque se había establecido que algunos elementos eran capaces de exhibir más de una valencia. Lothar Meyer señaló que la capacidad de saturación de los elementos (la valencia) aumenta y disminuye regularmente y uniformemente en ambos intervalos, por ejemplo:
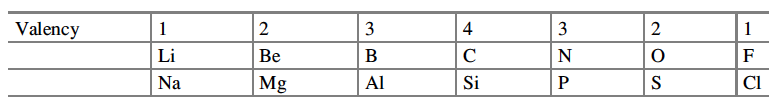
Así, a partir de un estudio de la dependencia periódica del comportamiento químico general de los pesos atómicos, surgió un nuevo conjunto de relaciones de valencia que, al menos durante los dos primeros períodos, revelaron una simplicidad subyacente que provocaría aún más preguntas fundamentales.
1.2 Descubrimiento de los gases nobles
Aunque la tabla periódica de Mendeleev condujo a muchas predicciones, no pudo anticipar por completo la existencia de un grupo completo de gases monoatómicos. El primero de los gases nobles descubierto por Lord Raleigh y William Ramsey en 1894 fue el argón. Además de no predecirse, las mediciones físicas en el argón sugirieron que era monoatómico, una propiedad que solo se había observado previamente para el vapor de mercurio. Como la valencia y el peso atómico eran los dos parámetros importantes para la tabla periódica, el peso atómico dependía de la atomicidad del nuevo elemento. Este problema se exacerbó cuando se dio cuenta de que la muestra de argón no se había obtenido en forma pura. Como el gas era completamente inerte, era necesario determinar su peso atómico a partir de mediciones de calor específicas, y una valencia de cero no tenía precedentes. En 1895, en una reunión en la Royal Society, Raleigh y Ramsey sugirieron que el nuevo elemento, si fuera un gas puro, tendría un peso atómico de 39.9, que no encajaría con la tabla periódica[18]. Sin embargo, si fuera una mezcla de dos gases con pesos atómicos de 37 (93.3%) y 82 (6.7%), los dos elementos encajarían perfectamente en posiciones entre cloro, potasio, bromo y rubidio. Reconociendo que este nuevo grupo de elementos puede representar una seria amenaza para su clasificación periódica, Mendeleev publicó su interpretación alternativa. Descartó la posibilidad de que fuera monoatómico porque no había espacio en la tabla periódica para tal elemento. Además, sería necesario tener un grupo de ocho en la tercera serie entre cloro y potasio. De hecho, concluyó que el nuevo gas era una forma triatómica de nitrógeno. En 1897 se descubrió helio terrestre y en 1900 criptón, neón y xenón, lo que confirma la presencia de una familia completamente nueva de elementos que no habían sido pronosticados por Mendeleev o cualquier otra persona. Ramsey propuso que sus pesos atómicos los colocaran entre los halógenos y los metales alcalinos, es decir, extendiendo la tabla de Mendeleev cada período en un elemento a la derecha[19]. Esto eliminó la amenaza que temía, y pudo celebrar en los siguientes términos “para mí es una confirmación gloriosa de la aplicabilidad general de la ley periódica”. Esta “magnífica supervivencia” del sistema periódico después de una “prueba crítica” había resultado. La incorporación de los gases nobles en la tabla periódica proporcionó un componente importante para el desarrollo de los principios de enlace químico propuestos por Lewis y Kossel en 1916[20].
1.3 Valencia
Los químicos y alquimistas anteriores a ellos, habían reconocido durante siglos que el comportamiento de las especies químicas estaba regido por un tipo de afinidad química, que resultaba de enlaces químicos específicos. En 1704, Sir Isaac Newton esbozó su famosa teoría de la vinculación atómica, en la “Consulta 31” de su Opticks, mediante la cual los átomos se unen entre sí por alguna “fuerza[21]”. Reconoció teorías previas sobre cómo se pensaba que los átomos se unían entre sí, es decir, “átomos enganchados”, “pegados por el reposo” o “pegados por movimientos conspiradores”, pero favoreció la idea de que la cohesión mediante la cual “las partículas se atraen entre sí por cierta fuerza, que en contacto inmediato es extremadamente fuerte, a pequeñas distancias realiza las operaciones químicas y llega no lejos de las partículas con ningún efecto sensible”.
El desarrollo de la valencia surgió de la teoría de la combinación química de Berzelius que enfatizaba el carácter electronegativo y electropositivo de la combinación de átomos[22]. A mediados del siglo XIX, Frankland, Kekulé, Couper, Butlerov y Kolbe, basándose en la teoría de los radicales, desarrollaron la teoría de la valencia en la que los elementos en los compuestos se unían mediante una atracción de polos positivos y negativos[23]. El concepto de valencia precedió al descubrimiento del electrón y la visión planetaria del átomo y puede rastrearlo hasta el artículo de 1850 por Frankland. Combinó las teorías más antiguas de los radicales libres y la “teoría de tipos” y demostró que los elementos tienden a combinarse con otros elementos para formar compuestos que contienen un número entero de elementos unidos, por ejemplo en los tres átomos unidos NH3, NI3, cuatro átomos unidos en CH4. Sobre la base de estos ejemplos y postulados, Frankland articuló la verdad: “Una tendencia o ley prevalece (aquí), y que no importa cuáles sean los caracteres de los átomos que se unen, el poder de combinación del elemento de atracción, si se me permite el término, siempre se satisface con el mismo número de átomos”. La convención de que los pares de átomos se mantienen unidos por una fuerza que se describió como un enlace fue utilizada por primera vez por Couper y Crum-Brown alrededor de 1860[24]. Representar un enlace por una línea eventualmente se convirtió en una convención gráfica de gran importancia para los químicos, pero por supuesto, no tiene realidad física directa.
La química tiene la habilidad de usar términos como valencia, electronegatividad y enlace que tienen una multiplicidad de significados. En su sentido más amplio, la valencia se ha utilizado para describir la capacidad de los elementos para combinarse con otros. Un enlace químico se define con mayor precisión como la fuerza que mantiene unidas a dos entidades químicas, pero la definición abarca una dualidad que en sus extremos se basa en enlaces electrostáticos (iónicos) o covalentes y entre una cantidad variable de carácter covalente e iónico. Este “poder de combinación” se describió posteriormente como cuantivalencia o valencia. La Unión Internacional de Química Pura y Aplicada (IUPAC) ha hecho varios intentos para llegar a una definición inequívoca de valencia[25]:
El número máximo de átomos univalentes (originalmente átomos de hidrógeno o cloro) que pueden combinarse con un átomo del elemento en consideración, o con un fragmento, o por el cual un átomo de este elemento puede ser sustituido.
Aunque la definición de Frankland funcionó bien para una amplia gama de moléculas inorgánicas y orgánicas, fue menos efectiva en la clasificación de sales. En estos compuestos, era más conveniente considerar el número de electrones que se transfieren entre los átomos. El “estado de oxidación” de un átomo en una molécula da la cantidad de electrones de valencia que ha ganado o perdido. A diferencia del número de valencia, el estado de oxidación puede ser positivo (para un átomo electropositivo) o negativo (para un átomo electronegativo). Por ejemplo, los estados de oxidación de los metales en NaCl, MgCl2 y AlCl3 son +1, +2 y +3, y el cloruro tiene una carga de -1. En Na2O, MgO y Al2O3, los estados de oxidación de los metales son idénticos a los de los cloruros porque el óxido tiene una carga de -2.
La clasificación periódica de Mendeleev y Meyer resaltó la relación entre la valencia de un elemento y su posición en un grupo particular de la tabla periódica. En 1904, Abegg amplió el concepto en una generalización que describió como el grupo de 8. Drude resumió claramente el grupo de 8 de Abegg de la siguiente manera: “El número de valencia positiva v de un elemento significa el número de electrones negativos sueltos en el átomo”; su número de valencia negativa v’ significa que el átomo tiene el poder de eliminar v’ electrones negativos de otros átomos, o al menos de unirlos más firmemente a sí mismo[26]. La perspectiva de que los electrones se relacionen con las valencias de los átomos siguió poco después del descubrimiento del electrón por Thomson, quien especuló que la valencia debe estar asociada con la transferencia de electrones entre los átomos. En los sólidos cristalinos, se especuló que las fuerzas que mantenían unidos los iones implicaban atracción electrostática entre cargas opuestas, pero estos conceptos no podían adaptarse fácilmente a los sólidos moleculares no polares. El estudio de Rutherford sobre la dispersión de partículas alfa por láminas de metal en 1911 mostró que, aunque la mayoría de las partículas pasaban directamente a través de la lámina, una pequeña cantidad se reflejaba en ángulos grandes. Estos experimentos llevaron a Rutherford a proponer un modelo de los átomos basado en una localización del núcleo en 1/10,000 del volumen ocupado por los electrones mucho más ligeros que ocupan el gran volumen del átomo. El estudio de Moseley en 1913 sobre las líneas espectrales de rayos X de los átomos mostró que su posición dependía principalmente del número atómico del átomo, es decir, el número de electrones o protones en un átomo neutro. Estas observaciones establecieron que la clasificación periódica de Mendeleev dependía principalmente del número atómico en lugar del peso atómico. Además, proporcionó una idea importante sobre su uso de la valencia como parámetro y sugirió que el número atómico debe estar relacionado con el número de electrones en un átomo de un elemento. Bohr desarrolló en 1913 una vista planetaria del átomo, que restringía el electrón a órbitas específicas basadas en la cuantificación del momento angular del electrón de acuerdo con la condición de Planck[27]. Bohr también reconoció que la estructura de la concha que resultó de sus restricciones cuánticas tenía implicaciones para comprender las estructuras electrónicas de las moléculas y la tabla periódica. Sommerfeld extendió el modelo a átomos más pesados ??y desarrolló un modelo basado en orbitales elípticos, que requirió dos enteros cuánticos[28].
1.4 Documentos de Lewis/Kossel
La visión moderna de la valencia se remonta principalmente a dos artículos publicados por Lewis y Kossel en 1916. Sus análisis independientes asociaron las estabilidades de los compuestos químicos de los elementos más ligeros al logro de ocho electrones en sus capas externas de electrones, es decir, el logro de configuraciones electrónicas de gas inerte.
Kossel centró su atención en el carácter fuertemente electropositivo de los elementos que suceden a los gases inertes y el carácter electronegativo de los elementos que preceden a los gases inertes[29]. Propuso que cuando los átomos de estos elementos se combinan, pierden o ganan electrones suficientes para alcanzar las capas cerradas asociadas con los átomos de gas inerte. Los iones positivos y negativos resultantes experimentan fuerzas de atracción electrostáticas clásicas y más que recuperar la energía gastada en formar los iones, especialmente si forman un sólido cristalino. Las cargas iónicas que resultan cuando los electrones se pierden o ganan pueden estar asociados con las valencias de los átomos. Por lo tanto, Kossel puede ser considerado como el co-creador de la regla del octeto, pero no reconoció la posibilidad de que los octetos también se puedan lograr compartiendo en lugar de la transferencia de electrones. Lewis propuso un análisis similar, pero también proporcionó una descripción de los enlaces químicos en compuestos moleculares orgánicos e inorgánicos. Propuso que también se puede lograr una configuración de gas inerte en una molécula como H2 si el par de electrones se comparte por igual entre ambos átomos, logrando así la misma configuración de capa cerrada que He. En la molécula de fluor F2, el intercambio de un par de electrones daría como resultado que ambos logren la misma configuración electrónica que un átomo de neón. Para Lewis, un par de electrones “compartido” resultó en un solo par de electrones que ocupan las capas de valencia de ambos átomos unidos. Postuló que en un enlace elemento-hidrógeno, el hidrógeno logró un doblete y el elemento al que estaba unido un octeto al compartir un par de electrones. Langmuir, que había sido estudiante de Lewis e hizo mucho para popularizar el modelo, introdujo el término enlace covalente para describir el intercambio de pares de electrones en tales moléculas para distinguirlo del enlace iónico o electrovalente encontrado en sales como Na+Cl[30]-.
Lewis no pudo explicar por qué dos electrones favorecían la formación localizada de enlaces de pares de electrones, aunque se esperaría que se repelan entre sí. De hecho, para resolver esta contradicción, propuso (erróneamente) que la ley de Coulomb podría no ser válida en las cortas distancias entre electrones encontradas en los enlaces. También reconoció la disparidad entre su visión estática de los electrones en los átomos y el modelo planetario que Bohr había desarrollado en 1913.
En 1923, Lewis propuso que si las órbitas de los electrones tenían una orientación espacial fija, entonces la posición promedio de los electrones coincidía con la posición fija de su par de electrones estáticos. El descubrimiento de que el electrón tenía un espín en 1925 y el desarrollo del principio de exclusión de Pauli llevaron al reconocimiento de que un par de electrones con el mismo espín se mantienen lo más separados posible, mientras que un par de electrones con espín opuesto redujeron la repulsión de electrones[31]. La importancia de estos efectos de correlación de carga y giro no se apreció completamente hasta la década de 1950 como resultado del trabajo de Lennard-Jones y Linnett[32].
1.5 Representación de las estructuras de Lewis
Las propuestas de Lewis y Kossel coincidieron con la estructura de la cubierta de los átomos que resultó del modelo híbrido clásico/cuántico para el átomo de hidrógeno desarrollado por Bohr y posteriormente extendido por Sommerfeld a otros átomos. No apreciaron completamente las implicaciones físicas de un modelo cuántico. Específicamente, Lewis basó su modelo en los siguientes postulados:
1. Los electrones del núcleo (o electrones centrales en capas cerradas) permanecen inalterados en todos los cambios químicos ordinarios.
2. Un átomo en una molécula tiende a contener un número par de electrones en sus capas de valencia.
3. Los electrones en los depósitos que se encuentran fuera del núcleo son mutuamente interpenetrables, y su emparejamiento conduce a la formación de un enlace covalente.
Lewis y Kossel sugirieron que los electrones en las moléculas y los iones forman grupos concéntricos de dos u ocho electrones, aunque los representaron de maneras muy diferentes. Lewis prefirió representarlos usando un modelo cúbico (su representación estática de los electrones condujo a una disposición simétrica si se ubicaban en los vértices de un cubo), mientras que Kossel prefirió usar anillos concéntricos para ilustrar las capas sucesivas. Las diferentes representaciones se resumen para neón. Lewis y Kossel concluyeron que las configuraciones electrónicas estables en las moléculas se parecen a los dos y ocho electrones encontrados en los gases inertes y observaron que el logro de estas configuraciones en las moléculas, ya sea compartiendo electrones o transfiriendo electrones, proporciona la fuerza impulsora para la unión química (Fig. 1).
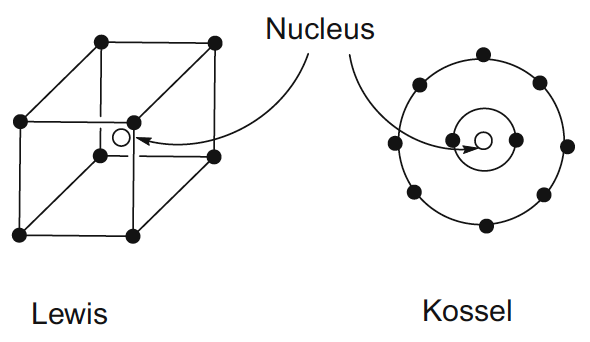
Fig. 1 Representaciones de electrones en átomos de Kossel y Lewis
Lewis había dado una conferencia sobre sus ideas en cursos de pregrado de 1902, es decir, en tiempos pre-cuánticos, pero se le desalentó de publicar el trabajo porque estaba incómodo con la dualidad de su teoría de enlaces químicos. También encontró problemático aplicar sus ideas a los hidrocarburos y especialmente a aquellos con enlaces múltiples. Como ha señalado, “no podía creer en dos tipos distintos de unión química”. Finalmente, en 1916, Lewis hizo la extensión importante para agregar una “regla de 2” a su “regla de 8”. Reconoció que con excepciones menores como NO, NO2 y ClO2, la gran mayoría de las moléculas, conocidas en ese momento, tenían incluso números de electrones. Por lo tanto, estableció la importancia del enlace par de electrones y reconoció que ya no pertenecía a ninguno de los átomos exclusivamente, sino que se compartía entre ellos. Extendió sus ideas a enlaces múltiples e inicialmente representó estos enlaces de pares de electrones gráficamente usando sus cubos como se muestra en la parte superior de la Fig.2.
Lewis señaló sus representaciones para las moléculas de hidrógeno y flúor y moléculas con dobles enlaces, por ejemplo eteno. No pudo representar los triples enlaces carbono-carbono que se encuentran en los alquinos usando la notación cúbica, y esto lo llevó a modificar el cubo a un tetraedro, en el cual pares de electrones se han atraído juntos (ver la parte inferior de la Fig. 2). El modelo combinaba dos ideas importantes: un par de electrones era responsable de cada enlace covalente, y las moléculas con enlaces simples, dobles y triples estaban representadas por un par de tetraedros que compartían vértices, bordes o caras. Este último incorporó las implicaciones estereoquímicas del átomo de carbono tetraédrico establecido anteriormente por van’t Hoff y LeBel. En publicaciones posteriores, Lewis abandonó las representaciones cúbicas y usó dos puntos para representar enlaces de pares de electrones y prefirió las estructuras de puntos que se muestran en la parte superior de la Fig. 3. Pedagógicamente, estas estructuras de puntos que enfatizan el logro del octeto de electrones alrededor del átomo central y los dobletes en hidrógeno todavía se utilizan para introducir conceptos básicos de enlace. Para enfatizar las valencias de los átomos, los orígenes de los electrones a veces se indican mediante el uso de los puntos y cruces que se muestran en la Fig. 3. A medida que los conceptos se vuelven familiares, las estructuras se representan mediante estructuras lineales. En química orgánica, esto también conlleva implicaciones al incorporar las estereoquímicas de los átomos de carbono, nitrógeno y oxígeno.
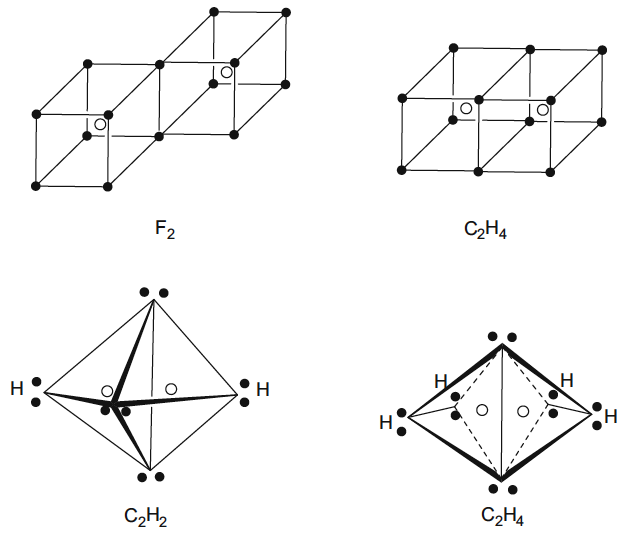
Fig. 2 Descripción de Lewis de los enlaces covalentes en F2 y C2H4 basados ??en el intercambio de electrones de dos cubos que conduce a enlaces simples y dobles respectivamente. El modelo no se pudo adaptar a C2H2, pero la descripción alternativa basada en pares de cuatro electrones en los vértices de un tetraedro podría dar como resultado el intercambio de tres pares de electrones necesarios para el triple enlace en C2H2.
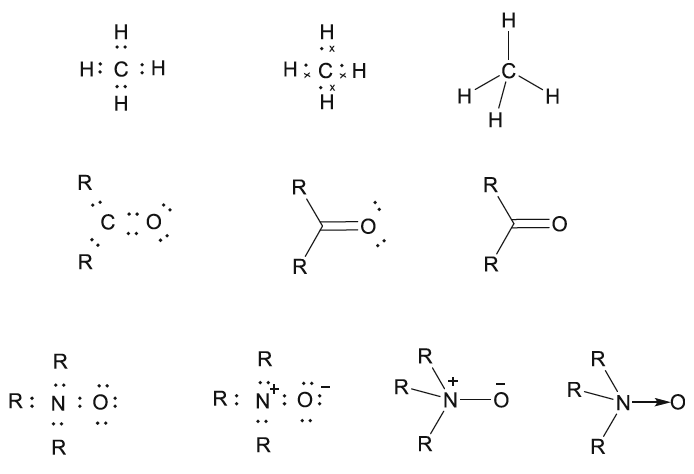
Fig. 3Representación de estructuras de Lewis basadas en el logro de capas cerradas mediante el intercambio de pares de electrones. Las estructuras de puntos iniciales se han reemplazado progresivamente por estructuras de líneas para representar los enlaces de dos centros y dos electrones. Los puntos solo se retienen cuando tienen consecuencias estereoquímicas o se requiere que representen reacciones orgánicas usando la notación de flecha rizada (Fig. 4)
1.6 Ácidos/Bases de Lewis: Representación del enlace dativo
En 1923, Lewis proporcionó una definición general importante de ácidos y bases: “Una sustancia ácida es aquella que puede emplear un par de electrones de otra molécula para completar el grupo estable de uno de sus propios átomos”. La teoría del ácido Brønsted-Lowry ácido-base fue publicada en el mismo año. Las dos teorías son distintas pero complementarias. Sin embargo, Lewis sugirió que un donante de pares de electrones se clasifique como base y un receptor de pares de electrones se clasifique como ácido. Langmuir reconoció que Me3BNH3 y Me3CCH3 eran isoelectrónicos y, en consecuencia, los enlaces B-N y C–C en sus centros deben estar estrechamente relacionados, ya que ambos se basan en el intercambio de un par de electrones. Sidgwick propuso que cuando ambos electrones provienen de uno de los átomos, podría describirse como un enlace covalente dativo o enlace coordinado. La distinción no fue aceptada universalmente, y Pauling, por ejemplo, rara vez usó los términos coordenadas o enlaces dativos en sus publicaciones y libros. Las representaciones alternativas de los enlaces covalentes dativos se muestran en la parte inferior de la Fig. 3. La teoría ácido/base de Lewis ha tenido un impacto importante en la comprensión de las reacciones de las moléculas orgánicas y Sidgwick la extendió a los compuestos de coordinación de metales de transición estudiados por Werner. Para representar las reacciones orgánicas como una serie de pasos ácido/base de Lewis, es común indicar los pares solitarios en las moléculas orgánicas como se muestra en la parte inferior de la Fig. 3. Ingold y Robinson fueron los principales responsables de mostrar cómo las ideas ácido/base de Lewis y las estructuras de Lewis podrían usarse para representar reacciones orgánicas, y la representación de flecha rizada resultante[33], que puede verse como una extensión del enlace dativo Sidgwick, es universalmente aceptada y utilizada para describir las rutas mecanicistas de las reacciones orgánicas. La Figura 4 ofrece algunos ejemplos específicos de la notación tal como se usa en la química orgánica actual.
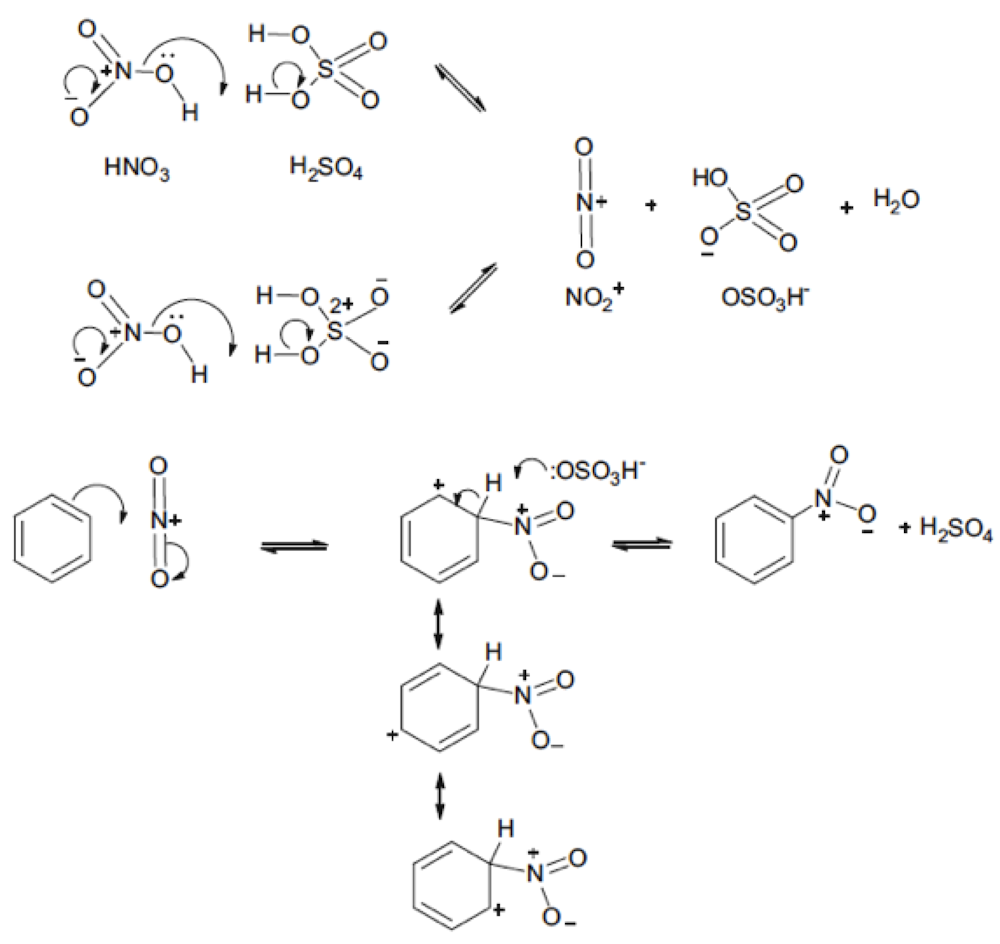
Fig. 4 Un ejemplo del uso de la notación de flecha rizada para representar el curso de las reacciones orgánicas. Las estructuras de resonancia que se muestran en el centro de la figura sugieren que los grupos liberadores de electrones en los sustituyentes orto y para del anillo de benceno favorecerán el proceso de sustitución. Aunque los reactivos que contienen azufre se dibujan con enlaces múltiples, la parte superior de la figura muestra que la notación de flecha rizada funciona igualmente bien si se dibujan estructuras de octeto de enlace único para estos compuestos
Robinson, Lapworth, Ingold, Pauling y Wheland ampliaron estos conceptos básicos para describir los efectos inductivos y mesoméricos de los sustituyentes en las moléculas orgánicas y proporcionaron una metodología muy ampliamente aceptada[34]. Esto explicaba las ubicaciones preferidas de las reacciones de sustitución en los anillos aromáticos y las tasas relativas de estas reacciones de sustitución. Un ejemplo específico se muestra en la Fig. 4. La notación de flecha rizada proporcionó una forma conveniente de describir la distribución de cargas en moléculas orgánicas y estados de transición. La adhesión de la regla del octeto asegura que el movimiento de un par de electrones desde un átomo (o enlace) solo se permite si se crea simultáneamente un agujero de pares de electrones para aceptarlo; esto define la ruta a través de la molécula. Esta conveniente notación fue apuntalada por el modelo de enlace de valencia desarrollado por Pauling y particularmente el concepto de resonancia. Sidgwick y Sutton proporcionaron evidencia experimental de estos efectos inductivos y mesoméricos midiendo los momentos dipolares de una amplia gama de moléculas e interpretaron los datos utilizando los modelos de enlace desarrollados por Pauling.
1.7 Resumen Lewis
Lewis ha ganado más reconocimiento por desarrollar un modelo de vinculación coherente que Kossel, pero al igual que Mendeleev, esto no fue reconocido por la concesión de un Premio Nobel, ¡aunque fue nominado más de 35 veces! En 1923, Lewis desarrolló los conceptos que habían sido presentados en el 1916 Journal of the American Chemical Society en su libro “Valence and the Structures of Atoms and Molecules”. Pauling reconoció su enorme contribución al dedicarle su clásica “Naturaleza del enlace químico” en 1938. En resumen, su teoría incorporó las siguientes ideas básicas:
1. La descripción del enlace químico depende de hacer una distinción entre los electrones de valencia, que contribuyen al enlace químico, y los electrones centrales, que no participan significativamente en el enlace químico.
2. Un enlace químico covalente resulta de compartir pares de electrones.
3. Un enlace iónico resulta de la transferencia de electrones del átomo electropositivo al átomo electronegativo. El número de electrones transferidos es dictado por el logro de una configuración de gas inerte.
4. La descripción de Lewis-Kossel proporcionó una descripción coherente del enlace químico, que depende del logro de la regla del gas inerte, ya sea compartiendo o transfiriendo electrones.
5. Las moléculas covalentes pueden tener pares de electrones involucrados en enlaces químico covalentes y también pares de electrones que no contribuyen al enlace químico. Por ejemplo, F2 tiene un enlace covalente que mantiene unidos los átomos de flúor y tres pares de electrones sin enlace en cada átomo de flúor.
6. Aunque las moléculas homonucleares como Cl2 y F2 son no polares, NaCl y KCl son altamente polares. Destacó la similitud entre muchos ácidos Brønsted, con la eliminación de compuestos moleculares y una distinción entre afinidades primarias y secundarias.
7. Proporcionó una notación efectiva de las estructuras electrónicas de moléculas inorgánicas y orgánicas. Inicialmente, esto se basó en la representación de pares de electrones como dos puntos, pero posteriormente se desarrolló de modo que los enlaces covalentes se representaban mediante líneas que unían los átomos y los pares de electrones no unidos como dos puntos.
8. Anticipó la electronegatividad como una forma de describir los enlaces polarizados, que cerró la brecha entre las formas extremas de enlace covalente e iónico.
9. Proporcionó una forma general de dar cuenta de las reactividades de los compuestos insaturados y el efecto de los sustituyentes en las regioselectividades de muchas reacciones orgánicas.
10. La definición del enlace químico como un par de electrones compartido podría extenderse para describir el enlace dativo y la elaboración de las interacciones ácido/base de Lewis.
Lo que es notable es el éxito y el uso generalizado de un modelo que los críticos severos dirían que se debe más a la numerología que a la física moderna y que no se basó sólidamente en la física cuántica o incluso en la de Newton. De manera contradictoria, define el enlace químico en términos de una interacción electrostática clásica entre iones con carga opuesta (el enlace iónico) y el emparejamiento de electrones cargados negativamente que comparten una pequeña región de la molécula (el enlace químico covalente). No es sorprendente que esta contradicción hizo que Lewis retrasara la publicación a partir de 1902 cuando presentó por primera vez las ideas básicas a los estudiantes universitarios en sus conferencias. La descripción moderna del enlace químico se basa en una descripción mecánica cuántica de átomos y moléculas que depende de definir el electrón en un átomo no como una partícula sino como una onda y cuyas propiedades dependen de cuatro números cuánticos, tres de los cuales definen el radial y características nodales de la onda y la cuarta el giro del electrón. La imagen orbital resultante del enlace químico no solo ha fomentado el desarrollo de representaciones pictóricas que explican la aparición de enlaces con órdenes de enlace que exceden los enlaces triples descritos por Lewis, sino que también ha proporcionado una gran comprensión de las geometrías tridimensionales de las moléculas y sus patrones de reactividad. Las generalizaciones de Lewis y Kossel no ayudaron a definir estas cuestiones fundamentales de la física, pero enfatizaron la importancia del par de electrones en un enlace químico y la importancia de lograr configuraciones de gas inerte en iones y moléculas. Lo más importante es que proporcionó un medio muy efectivo de comunicar en la comunidad química la valencia, la estereoquímica de los átomos en las moléculas y una forma de auditar el movimiento de los pares de electrones entre los reactivos y los productos en las reacciones químicas.
Los químicos reconocieron desde una etapa temprana que el enfoque de Lewis-Kossel proporcionaba estructuras moleculares alternativas para moléculas con el mismo número de electrones de valencia. Esta ambigüedad fue incluso evidente para los elementos que pertenecen al mismo grupo de la tabla periódica. Por ejemplo, aunque N2 y O2 son moléculas diatómicas que tienen fuertes enlaces múltiples, los elementos relacionados fósforo y azufre tienen formas alotrópicas, que se basan en enlaces simples entre los elementos. El número de enlaces covalentes formados por cada átomo es idéntico, pero los elementos más ligeros muestran una gran preferencia por formar enlaces múltiples como se muestra en la figura 5.
La capacidad de la primera fila larga de elementos para formar enlaces múltiples fuertes es una característica general importante de la tabla periódica, pero la descripción clásica de Lewis del eteno tiene que modificarse para los compuestos análogos de los elementos del Grupo 14 más pesados. Como se muestra en la Fig. 6, la estructura plana característica del eteno ya no se mantiene y las moléculas muestran una estructura plegada, y el ángulo de plegado aumenta con el número atómico del elemento. Es de destacar que la estructura resultante puede describirse como estructuras singulares de “carbonoide” que puede considerarse que interactúa más débilmente a través de enlaces dativos como se muestra en la Fig. 6.
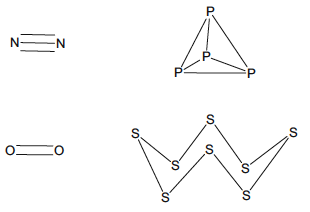
Fig. 5 Estructuras de Lewis que dan lugar a atenuadores de múltiples enlaces o compuestos poliédricos y anulares para elementos que pertenecen al mismo grupo de la tabla periódica
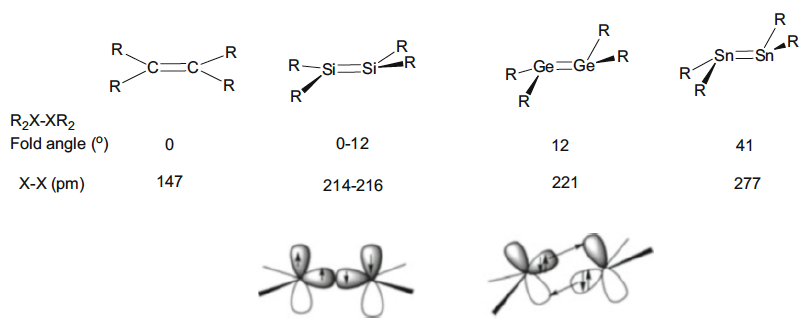
Fig. 6 Dos pares de electrones que forman un doble enlace en eteno usando la descripción clásica de Lewis o dos enlaces dativos. Este último se observa en análogos de eteno para los elementos del grupo 14 más pesados. Las consecuencias geométricas de los diferentes modos de enlace dan como resultado el plegamiento progresivo de la molécula
1.8 Extensiones del modelo Lewis/Kossel
1.8.1 Las generalizaciones de las estructuras de Lewis
La discusión anterior ha indicado algunas de las limitaciones de la descripción original de Lewis/Kossel del enlace químico y la manera en que se ha adaptado para asimilar la multitud de nuevos compuestos que han sido reportados de laboratorios químicos durante el último siglo. Central para el modelo es la definición del enlace químico como un par de electrones y la adherencia a la regla de octeto.
Langmuir, que desarrolló fórmulas específicas que relacionan la convalecia del átomo central con el número de electrones de valencia en los gases inertes, amplió la relevancia de las capas electrónicas completas asociadas con los gases inertes. Dado que los números atómicos de los gases inertes son 2, 10, 18, 36, 54 y 86, estos números se identificaron con la finalización de configuraciones electrónicas estables. Si se excluyen los electrones centrales, estas configuraciones se asocian con 2, 8, 8, 18, 18, 32 electrones de valencia. Bury aclaró la propuesta de Langmuir al sugerir que el número máximo de electrones en las diversas capas es de 2, 8, 18 y 32[35]. Bury señaló que en los átomos de metales de transición y lantánidos, se produce la construcción interna, es decir, el llenado de las capas electrónicas internas, mientras que los más externos permanecen constantes. Estos desarrollos llevaron a los químicos a usar la regla del octeto para las moléculas orgánicas y del grupo principal, y Blanchard[36] aplicó la regla de los 18 electrones a los complejos de carbonilo de metales de transición como el Ni(CO)4. Sidgwick introdujo en 1923 un procedimiento alternativo de recuento de electrones, basado en las estructuras de capa de electrones propuestas por Bohr y Bury[37]. La regla del número atómico efectivo (EAN), se centró no solo en el recuento de electrones de capa de valencia, sino en la cuenta total del electrón del átomo. El logro de un octeto o una configuración externa de 18 electrones era equivalente a alcanzar el recuento total de electrones (o número atómico) del gas noble más cercano. La regla EAN de Sidgwick se aplicó por primera vez al creciente número de carbonilos y nitrosilos de metales de transición por Reiff en 1931[38], y en 1934 Sidgwick extendió su uso a complejos con puentes, carbonilos. Sidgwick y Blanchard popularizaron la regla en la década de 1940. En la década de 1960, hubo una reversión a los procedimientos de conteo de electrones basados ??únicamente en los electrones de valencia, porque las moléculas del grupo principal podían referirse a la regla del octeto, y las tres filas de metales de transición podían referirse a la regla de los 18 electrones. La regla EAN de Sidgwick, que incluye los electrones centrales químicamente inactivos, da como resultado un recuento de electrones separado para cada fila del grupo principal y bloques de metal de transición. El octeto y las reglas de 18 electrones están sujetas a muchas excepciones, pero, sin embargo, demostraron ser muy útiles como herramienta pedagógica en la química organometálica e inorgánica[39].
Se señaló anteriormente que la regla del octeto inicial se extendió a una regla de 18 electrones para compuestos de metales de transición, y la notación de enlace dativo introducida por Sidgwick se usó muy ampliamente para describir compuestos de coordinación y compuestos organometálicos. La dualidad que surge de la descripción formal del enlace en dichos compuestos en términos de estados de oxidación formales del ion metálico central o un modelo covalente basado en la valencia del metal ha presentado ciertos problemas, que se han discutido con cierta extensión en las revisiones de Green y Parkin[40]. El creciente número de compuestos organometálicos desde 1950 y su importancia como intermedios en procesos catalíticos condujeron a un estudio detallado de los complejos de alquenos y carbonilos de metales de transición en estados de baja oxidación. Esto reveló que el enlace dativo en tales compuestos podría proceder simultáneamente en ambas direcciones, es decir, desde un par de ligando solitario al metal y desde un orbital d lleno en el metal a un orbital vacío en el ligando. Este modelo de enlace sinérgico representa uno de los resultados más importantes del modelo de pares de electrones de Lewis, Green y Parkin han introducido una notación conveniente y flexible para clasificar tales compuestos.
En esta revisión, se dirigirá la atención hacia algunas diferencias importantes en la forma en que los compuestos del octeto y 18 electrones se representan comúnmente en la literatura para describir estructuras y reacciones. La Figura 7 compara las representaciones de los compuestos típicos del grupo principal y los metales de transición que se ajustan al octeto y las reglas de 18 electrones y enfatiza la notación de enlace dativo introducida por Sidgwick. La presencia omnipresente de CO como ligando donador de dos electrones resultó en una simplificación, por lo que la flecha de enlace dativo se reemplaza comúnmente por una sola línea de enlace, aunque esto puede ser engañoso para los recién llegados al campo, que han sido introducidos en las reacciones ácido-base de Lewis representadas por flechas de enlace dativo. La otra omisión importante se refiere a los pares solitarios. En los compuestos de octeto, los pares solitarios se muestran claramente y son importantes para el uso de estas fórmulas de Lewis para describir las reacciones de estas moléculas a través de las representaciones de flechas rizadas. También tiene implicaciones estructurales porque estos pares solitarios son estereoquímicamente activos y ocupan espacio como si fueran enlaces covalentes. Por lo tanto, las tres moléculas del grupo principal en la Fig. 7 pueden estar relacionadas con el tetraedro original con pares solitarios que reemplazan sucesivamente los enlaces. Sidgwick y Powell reconocieron la importancia estereoquímica de los pares solitarios en las moléculas del grupo principal y la revisaron en 1940[41]. Esta generalización estereoquímica que se describió como la teoría del par de electrones de la capa de valencia fue amplificada posteriormente por Gillespie y Nyholm[42]. Para los carbonilos de metales de transición, los metales también tienen pares de electrones que no se usan en los enlaces sigma metal-ligando, pero generalmente no se muestran en las representaciones de Lewis/Sidgwick. Específicamente, Cr, Fe y Ni tienen 6, 8 y 10 electrones emparejados en el metal, es decir, 3, 4 y 5 pares de electrones, que se omiten (Fig.7). Esta diferencia puede haber surgido inicialmente por motivos estéticos y de impresión, pero también refleja la opinión actual de que los electrones de valencia pertenecen a una capa interna. Mostrar todos estos pares de electrones puede conducir a representaciones bastante abarrotadas, como se muestra en la parte inferior de la Fig. 7, y más significativamente, los pares de electrones no son estereoquímicamente activos en la forma descrita anteriormente para los compuestos de octeto. Esta diferencia significativa se ha interpretado utilizando una base mecánica cuántica del electrón libre que se describe como El Modelo de Densidad de Electrones Esférico Complementario[43]. Los ligandos y los orbitales metálicos en un compuesto de 18 electrones están relacionados con los de un gas inerte, y sus representaciones de función de onda proporcionan conjuntos ortogonales completos y complementarios. Las características nodales de los pares de electrones localizados en el metal los hacen ortogonales a los orbitales metal-ligando y, en consecuencia, no son estereoquímicamente activos. Zhenyang Lin ha discutido este aspecto de la química de coordinación del metal de transición en un capítulo separado de este volumen[44].
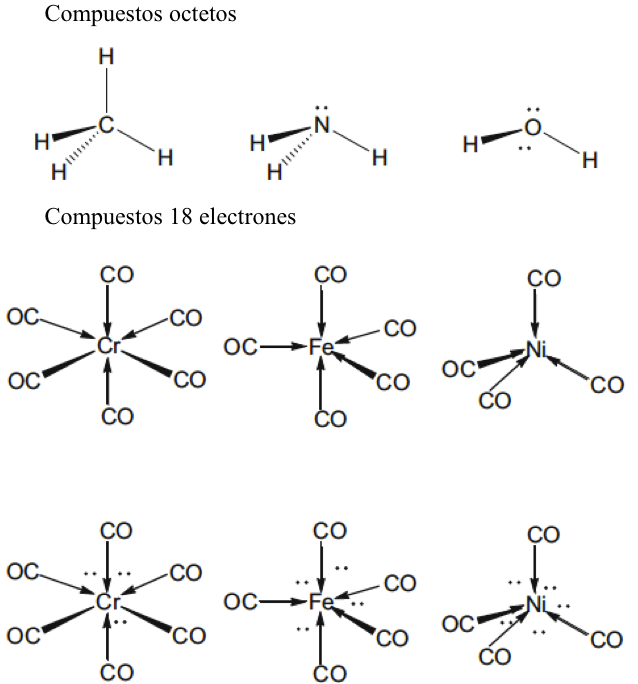
Fig. 7 Comparación de las estructuras de Lewis para el grupo principal típico y los compuestos de metales de transición.
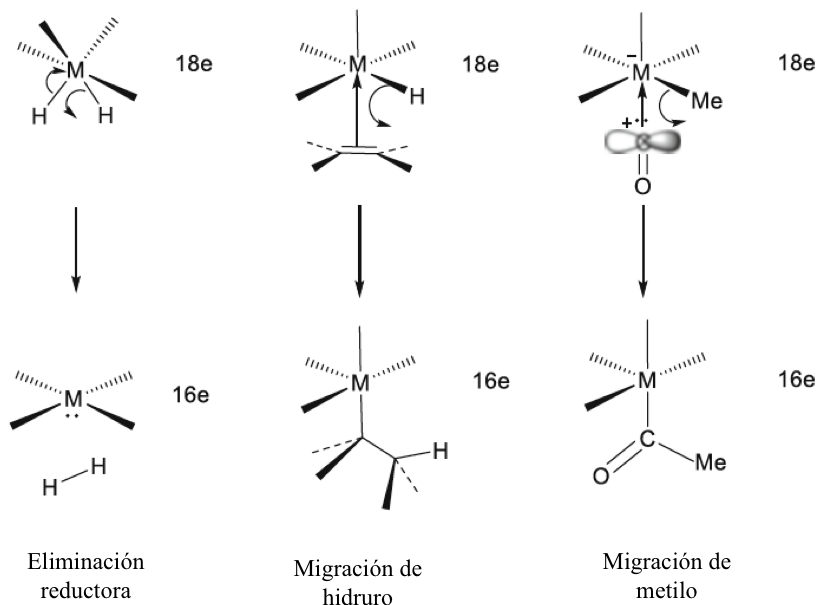
Fig. 8Representaciones de flechas rizadas de transformaciones organometálicas arquetípicas.
La otra pregunta intrigante que resulta de la extensión de la notación de Lewis a la química del metal de transición es por qué la notación de flecha rizada que se usa tan comúnmente en la química orgánica no se ha usado más ampliamente en la química organometálica y de coordinación[45]. La Figura 8 ilustra las transformaciones primarias de complejos metálicos, a saber, adición oxidativa, migración de hidrógeno y migración de metilo usando notación de flecha rizada que tiene análogos en química orgánica. Es de destacar que todas las transformaciones y sus reacciones inversas implican cambios en los recuentos de electrones de 18 a 16 o viceversa, y en consecuencia el mantenimiento de la reserva de electrones es ligeramente más complejo que el de las reacciones orgánicas donde se mantiene la regla del octeto. El uso insignificante de esta notación en contraste con la química orgánica puede resultar, porque, en contraste con las reacciones orgánicas, hay un pequeño número de centros involucrados. Sin embargo, Ghosh y Berg han demostrado recientemente cómo la notación de flecha rizada se puede utilizar para sistematizar la química del grupo principal[46]. Los movimientos concertados de pares de electrones a lo largo de muchos centros y alrededor de un anillo de átomos son mucho menos comunes en las reacciones de metales de transición. La representación de π-enlaces ligandos con ligandos de carbono 3–8 en complejos organometálicos proporciona una complicación adicional para este tipo de representación. Para organometálicos simples, estos problemas pueden superarse utilizando formas canónicas como las ilustradas en la figura 9.
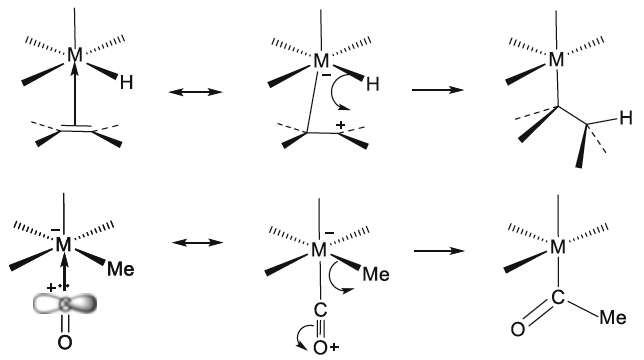
Fig. 9Formas canónicas que ilustran cómo se puede usar la notación de flecha rizada para compuestos organometálicos.
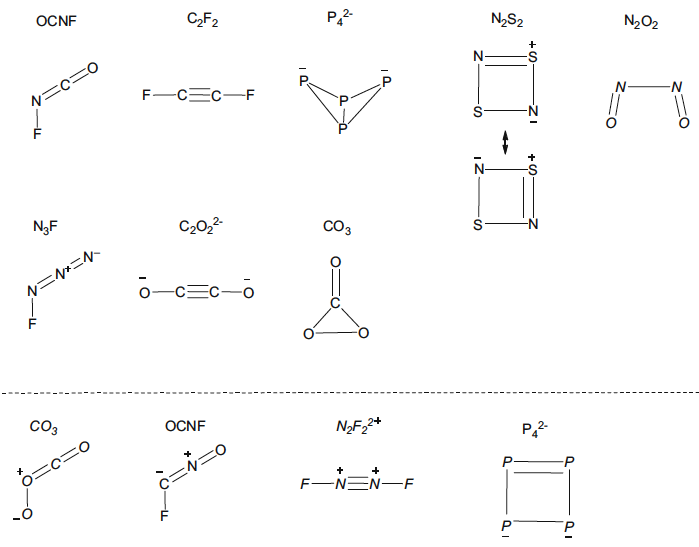
Fig. 10 Estructuras alternativas para iones y moléculas con 4 átomos y 22 electrones de valencia. Dado que las estructuras de Lewis se basan en el logro de la configuración de gas inerte en cada átomo, todas las estructuras tienen cinco enlaces. Las estructuras debajo de la línea son generalmente menos favorecidas debido a la naturaleza dipolar de las estructuras o la aparición de cargas idénticas en los átomos adyacentes.
1.8.2 Relaciones isoméricas e isoeléctronicas
El formalismo de la regla par-electrón/octeto muestra limitaciones significativas cuando se aplica a moléculas más complejas, porque se pueden escribir estructuras isoméricas alternativas, todas las cuales son consistentes con los supuestos de Lewis. Por ejemplo, las moléculas e iones con cuatro átomos y un total de 22 electrones de valencia son consistentes con todas las estructuras de Lewis que se muestran en la figura 10. Se producen estructuras lineales, angulares, de anillo y de mariposa, aunque cada estructura está asociada con cinco enlaces covalentes de dos electrones.
En consecuencia, se requieren criterios adicionales para establecer qué posibilidades son más estables. Los siguientes criterios proporcionan una forma preliminar de entender por qué se prefieren ciertas estructuras.
1. El átomo menos electronegativo generalmente se encuentra en ubicaciones centrales y los átomos más electronegativos en el exterior.
2. Los elementos más pesados ??favorecen las estructuras de anillo en lugar de estructuras lineales con enlaces múltiples.
3. Las estructuras con átomos no cargados generalmente se prefieren en relación con las estructuras cargadas, y los átomos con carga positiva generalmente se desfavorecen para átomos muy electronegativos como F y O.
4. Las estructuras con la misma carga en los átomos adyacentes están desfavorecidas.
Las estructuras que se muestran sobre la línea punteada ilustran las implicaciones de estos criterios y sugieren estructuras más estables. Además, aunque la estructura que se muestra para N2O2 es consistente con el formalismo de Lewis, el enlace simple N-N débil significa que la estructura solo se observa a bajas temperaturas. La energía de disociación del dímero de NO es solo de 8.3 kJ mol^-1, y representa un ejemplo de una molécula que no está adecuadamente representada por los cálculos orbitales moleculares de Hartree-Fock. Las estructuras debajo de la línea punteada están desfavorecidas debido a que son dipolares o tienen cargas idénticas en los átomos adyacentes.
El número de enlaces, x, asociado con una estructura de Lewis puede resumirse mediante la siguiente relación, donde el número total de electrones de valencia, t, h y m representa el número de átomos de hidrógeno y del grupo principal:
donde h es el número de átomos de hidrógeno (EAN = 2, correspondiente a la capa cerrada para He) y m es el número de átomos del bloque p (que alcanzan los ocho electrones asociados con el gas inerte adyacente). Claramente, los ejemplos en la Fig. 10 comparten cinco enlaces en común debido a esta relación. Algunos otros ejemplos de esta relación se dan en la Tabla 1, y la Fig. 11 ilustra cómo x varía sistemáticamente a medida que varía el número de electrones de valencia t. A medida que aumenta t, el número de enlaces covalentes disminuye. La tabla proporciona ejemplos de moléculas precisas de electrones que obedecen la regla del octeto y también moléculas que aparentemente son hipovalentes (es decir, no alcanzan la regla del octeto) e hipervalentes (es decir, exceden la regla del octeto).
Tabla 1 Ejemplos de moléculas simples que se adhieren a la regla del número atómico efectivo (EAN)
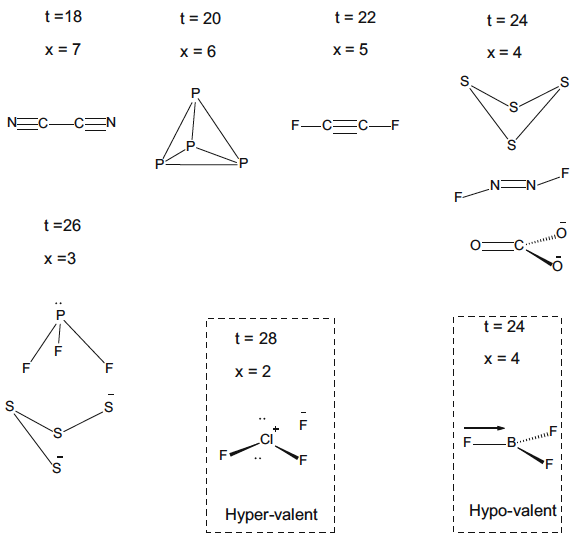
Fig. 11 Ejemplos de moléculas con el mismo número de átomos, pero con diferentes números de electrones de valencia, t, que resultan en una variación en el número de enlaces, x, para satisfacer los requisitos de la regla del octeto.
1.8.3 Principal grupo de moléculas Hipo-valencia e Híper-valencia
La Tabla 1 da ejemplos de moléculas precisas de electrones donde se obedece la regla del gas inerte. El número de enlaces es consistente con las estructuras determinadas. Estas moléculas pueden tener enlaces múltiples o enlaces simples. La tabla también da ejemplos de moléculas que no obedecen la regla del octeto, y el número previsto de enlaces no es consistente con x número de enlaces-σ. Dichas moléculas se describen como hipovalente o hipervalente según si la regla del octeto no se cumple o se excede[47]. En estas moléculas, si el valor x excede el número de enlaces-σ, indica el número de enlaces adicionales necesarios para hacer que el compuesto se ajuste a la regla del gas inerte. Por ejemplo, en BF3 y BH3, x = 4 si se obedece la regla del gas inerte, aunque solo hay tres enlaces B–F o B–H. Esta deficiencia puede compensarse agregando un enlace dativo como se muestra en la Fig. 12. En BF3 se ha propuesto una interacción mesomérica que involucra un orbital pπ en F perpendicular al plano y el orbital B 2p vacío. En BH3 esto está excluido, pero un enlace dativo intermolecular de un enlace B-H al orbital de boro 2p ayuda a resolver la hipovalencia. La resonancia entre las formas canónicas relacionadas con la simetría conduce a la estructura observada de B2H6, que se describe más comúnmente en términos de un par de enlaces B–H–B de tres centros y dos electrones[48]. Por supuesto, las restricciones estéricas que impiden la polimerización significan que esta forma de lograr la regla del octeto puede no ser realizable.
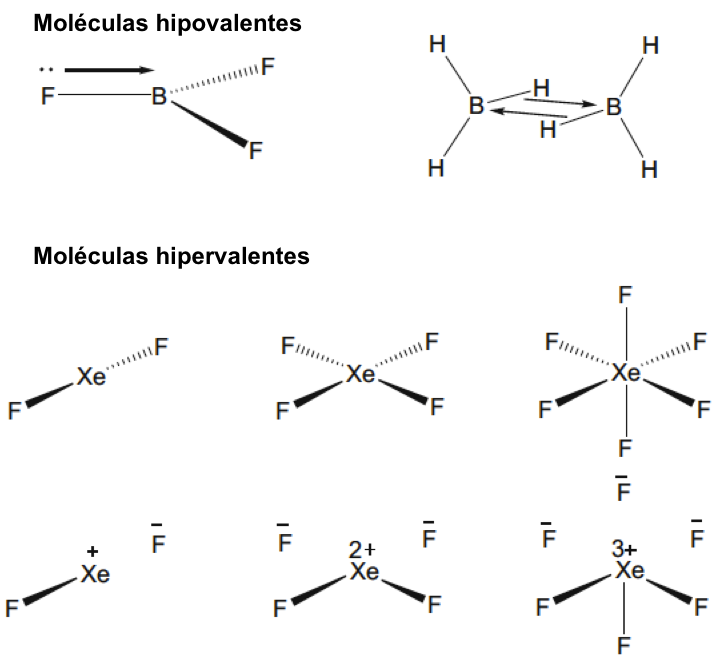
Fig. 12 La extensión de las ideas de Lewis para moléculas hipovalentes e hipervalentes. En el primero, la deficiencia de electrones se alivia mediante la formación de enlaces dativos, ya sea a partir de pares solitarios o enlaces B–H. En este último, la hipervalencia se alinea con la regla EAN utilizando estructuras de enlace de Lewis donde el átomo central tiene cargas positivas que compensan las cargas negativas en los flúor. Los resultados en la Tabla 1 sugieren que x está asociado con el número de enlaces Xe-F de tres centros y cuatro electrones en estas estructuras.
La configuración de gas inerte de capa cerrada ns^2np^6 para los gases nobles está asociada con un alto potencial de ionización y una afinidad electrónica desfavorable. Sus configuraciones electrónicas cerradas dan como resultado que el giro total y los momentos angulares orbitales sean cero y, por lo tanto, reducen la posibilidad de interacciones de pares de electrones con electrones en otros átomos. Por lo tanto, los gases nobles deberían proporcionar ejemplos robustos de la regla del octeto al no formar compuestos. De hecho, se podría argumentar que las ideas de Lewis pueden haber obstaculizado el descubrimiento de compuestos de gases nobles. El trabajo pionero de Bartlett sobre compuestos de xenón a principios de la década de 1960 mostró que los gases nobles más pesados ??forman compuestos con átomos electronegativos como F y O[49]. Sus electrones de valencia se involucran en enlaces de pares de electrones con átomos electronegativos como F y O cuando la energía de ionización del gas inerte es suficientemente baja. Para estos compuestos hipervalentes (XeF2, XeF4 y XeF6), x = 1, 2 y 3 de acuerdo con la fórmula dada anteriormente, es decir, la mitad del número de enlaces Xe-F.
Inicialmente, se pensaba que estos compuestos hipervalentes eran el resultado de la promoción de electrones de los orbitales de valencia de xenón en orbitales d superiores y la posterior formación de enlaces de dos electrones de dos centros con O o F, es decir, superan la regla del octeto. Las interpretaciones contemporáneas del enlace en estos compuestos favorecen la formación de enlaces de tres centros y cuatro electrones. Esta interpretación da como resultado las formas canónicas iónicas ilustradas en la Fig. 12, que cuando están en resonancia reproducen las geometrías simétricas observadas. En estas moléculas, x representa el número de enlaces de cuatro electrones y tres centros en la molécula.
Estos ejemplos ilustran la forma en que los químicos han modificado las representaciones formales de enlaces Lewis de dos electrones y dos centros para extender la descripción a compuestos hipo e hipervalentes. Implica una extensión de las ideas para abarcar el intercambio de pares de electrones entre tres en lugar de dos átomos y el uso de resonancia para hacer coincidir las formas canónicas con la simetría de la molécula. Para los compuestos hipo e hipervalentes, la regla del octeto se puede lograr formando enlaces dativos multicéntricos o complementarios.
Como observó Moeller en la década de 1950, “aunque la regla del octeto es definitivamente un concepto útil, sus aplicaciones son limitadas y no debería recibir la atención universal que normalmente se enfoca en ella. Es mucho más relevante que la atención se dirija al importante fenómeno del emparejamiento de electrones. El concepto de que los electrones buscan emparejarse entre sí es casi universal en su aplicación y siempre es útil como primera aproximación para predecir el comportamiento químico. Esta “regla de dos” es mucho más fundamental que la “regla de ocho”. Para las sensibilidades contemporáneas, esto parece un poco duro y generalmente multicéntrico, y los vínculos dativos que favorecen la adhesión a la regla del octeto representan un punto de partida conveniente para la discusión.
1.8.4 Relaciones isoeléctronicas
Aunque los ejemplos en la sección anterior han llamado la atención sobre la posibilidad de que las moléculas e iones con el mismo número de átomos y electrones de valencia puedan tener diferentes estructuras, la relación isoelectrónica ha demostrado ser una forma importante de conectar moléculas con grupos similares de átomos. La regla EAN o la regla equivalente basada en el número de electrones en las capas externas de los gases inertes enfatizó las siguientes relaciones isoelectrónicas que resultaron ser particularmente útiles para interrelacionar las estequiometrías y las estructuras de las sales inorgánicas[50]:
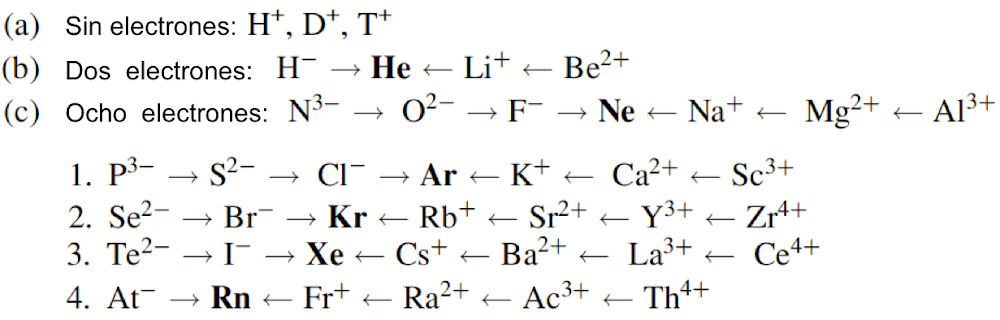
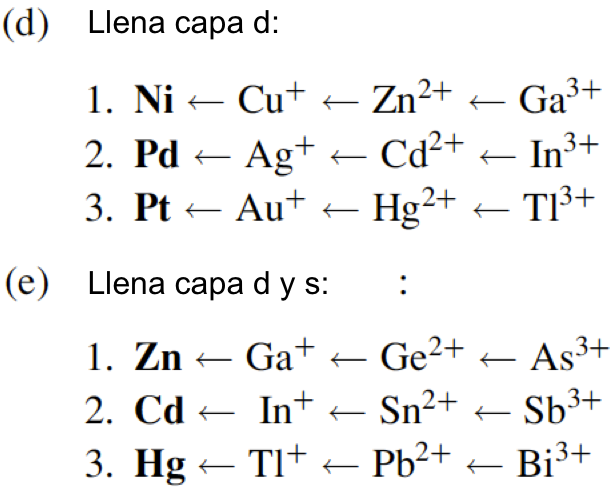
También destacó las relaciones entre los iones que no confirmaron la regla EAN, específicamente las siguientes que tenían una capa d completa, pero las capas s y p vacías (d) o una capa s completa, pero una capa p vacía (e). Los iones mostrados en (e) fueron descritos por Sidgwick como compuestos de pares inertes.
Estas relaciones isoelectrónicas han dado como resultado en los últimos años compuestos que contienen cationes de metales alcalinos, por ejemplo Na+, K+, etc., y el aislamiento de sales del anión aurida Au. Son análogos del anión hidruro. A medida que avanzaba el estudio de los compuestos de metal de transición y lantánido, estas ideas se extendieron a los iones metálicos que tenían capas semillenas, es decir, Mn^2+, Eu^2+, Tb^4+, con cada uno de los orbitales d o f que contienen un solo electrón y todos los electrones tienen giros paralelos.
En 1919 Langmuir observó que las moléculas y los iones que contenían el mismo número de átomos y el mismo número total de electrones invariablemente tenían estructuras idénticas. Describió tales series como grupos isostéricos y la Tabla 2 a continuación proporciona ejemplos específicos.
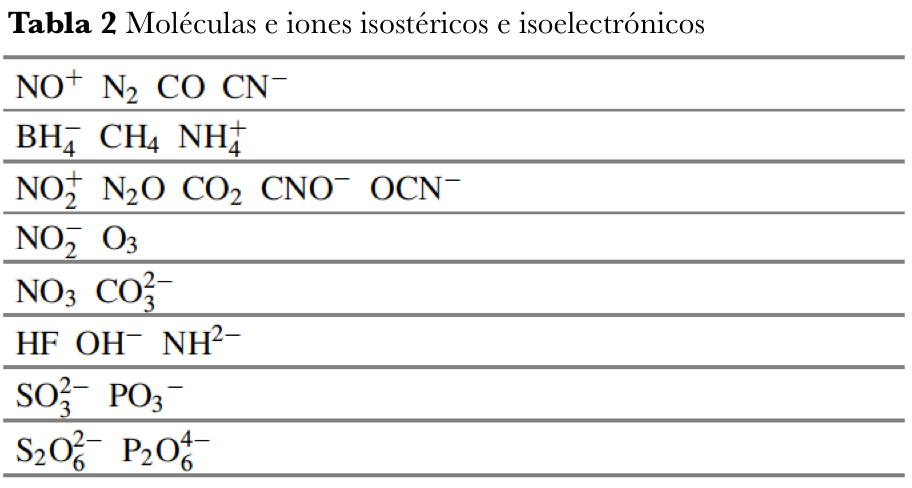
Los isómeros y las relaciones isoelectrónicas estrechamente relacionadas todavía son ampliamente utilizados por los químicos inorgánicos como un predictor efectivo de nuevas moléculas. Estás relaciones isoelectrónicas proporcionan una buena guía para la aparición y las estructuras de las moléculas predichas, aunque la variación en las cargas de los iones puede influir en sus propiedades ácido/base de Lewis y sus propiedades redox. La tabla 3 proporciona ejemplos adicionales de compuestos inorgánicos del grupo principal, que tienen el mismo número de electrones de valencia y geometrías similares.
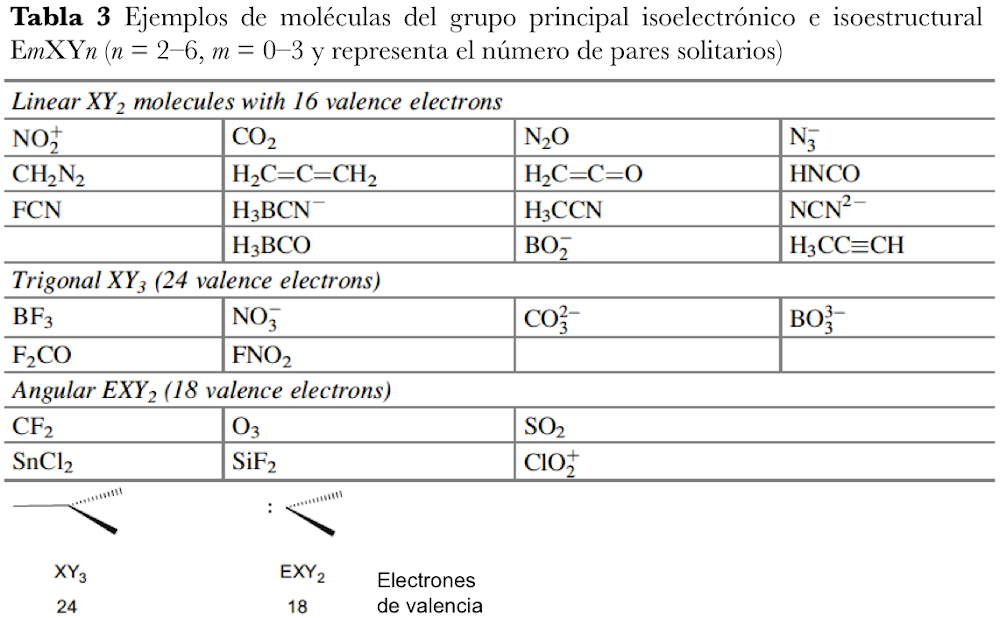
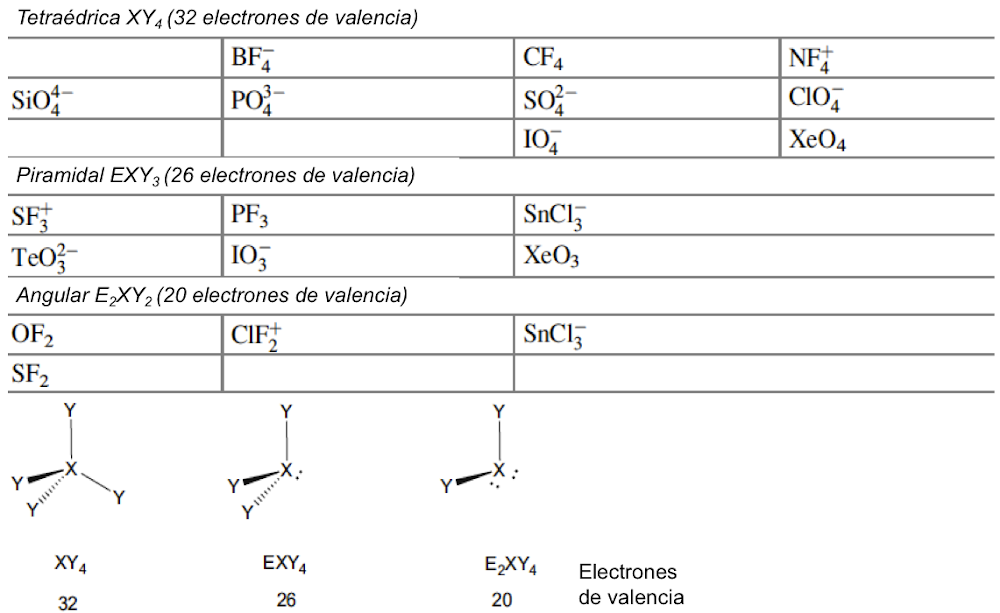
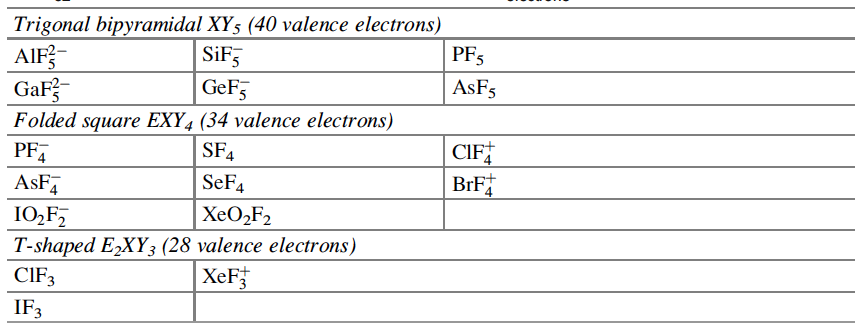
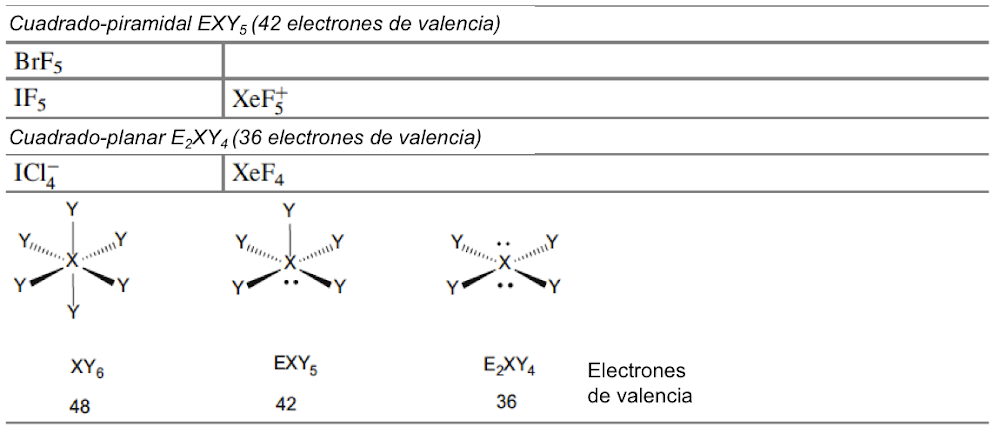
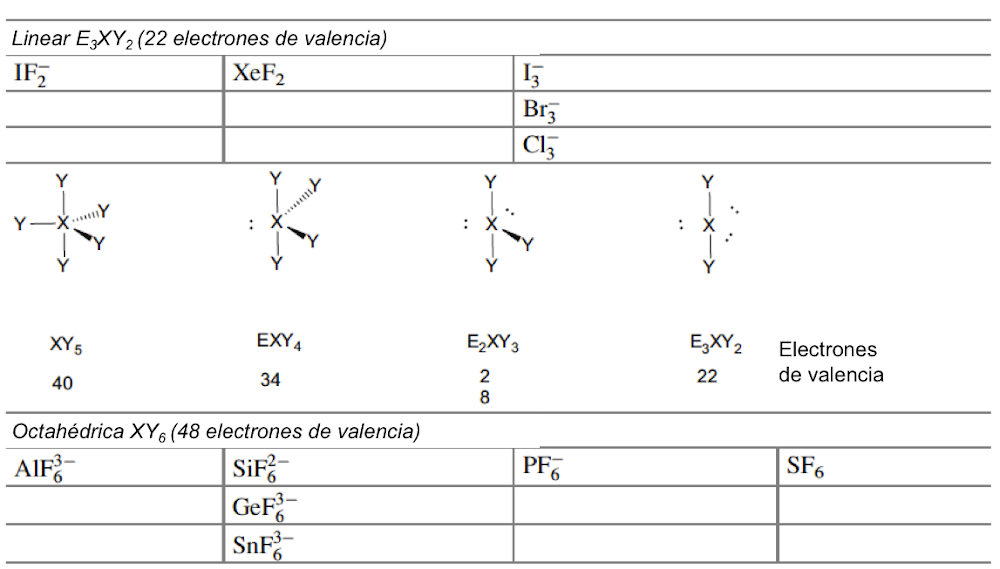
Estas relaciones isoelectrónicas desempeñaron un papel importante en el desarrollo de la teoría de la repulsión del par de electrones de la capa de valencia (VSEPR) y también en la aceptación más general de los modelos estereoquímicos basados ??en modelos orbitales moleculares. Dentro del marco de la teoría de los orbitales moleculares, dichos orbitales deslocalizados definidos por consideraciones de simetría se calculan y luego se llenan utilizando el principio de aufbau de manera análoga a la desarrollada para los átomos de polielectrones. De ello se deduce que el mismo modelo básico se puede utilizar para tener en cuenta las propiedades químicas y espectroscópicas de la serie isoestructural de moléculas o iones. Para una serie isoelectrónica e isoestructural, XYn, los mismos orbitales moleculares de enlace están ocupados, pero el grado de localización en los átomos centrales y periféricos reflejará la diferencia de electronegatividad entre X y Y. Walsh fue particularmente influyente en la introducción del análisis orbital molecular en estas moléculas. Los diagramas de Walsh también fueron importantes para resaltar las diferencias en la geometría entre los estados fundamental y excitado y el seguimiento de cómo el aumento en el número de pares de electrones puede cambiar como resultado del aumento del número de electrones.
Tabla 4 Diferencias de electronegatividad
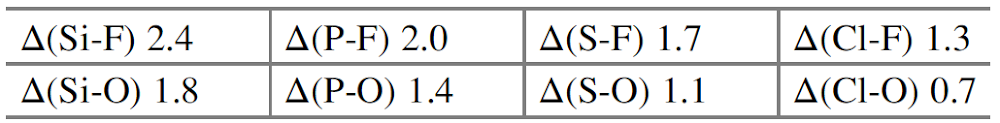
La naturaleza deslocalizada de los orbitales moleculares elimina la fuerte conexión inherente a las estructuras de Lewis y al método de enlace de valencia en las órdenes de enlace formales. Por ejemplo, la serie de moléculas tetraédricas SiF4, POF3, SO2F2, ClO3F, SiO44-, PO43-SO42- y ClO4- son isoelectrónicos y tienen 32 electrones de valencia (x = 4 de acuerdo con la fórmula presentada anteriormente); el reemplazo de flúor por oxígeno sugiere una mayor formación de enlaces múltiples de la manera familiar para los químicos orgánicos, pero dentro del marco orbital molecular, todas las moléculas tienen los mismos orbitales moleculares ocupados. La extensión de la localización en el átomo central cambia de acuerdo con la diferencia de electronegatividad entre el átomo central y los átomos periféricos (Tabla 4). Además, las simetrías de estas moléculas no proporcionan una distinción clara entre los orbitales σ y π de una manera que sea familiar para los químicos orgánicos, y solo los cálculos orbitales moleculares precisos pueden proporcionar una indicación de las polaridades de enlace, y de hecho, la contribución realizada por los orbitales 3d en el átomo central.
Aunque las relaciones isoelectrónicas e isostéricas generalmente se atribuyen a Langmuir, Grim y Erlenmeyer las extendieron. Grimm consideró todas las moléculas con el mismo número de electrones de valencia independientemente del número de átomos involucrados y utilizó el isomorfismo como criterio. Erlenmeyer concluyó que solo se debería contar el número externo de electrones al proponer el isoesterismo y lo aplicó ampliamente a las moléculas orgánicas.
1.8.5 Teoría de los electrones pares de repulsión de la capa de valencia
La observación de que los pares solitarios en las moléculas del grupo principal son estereoquímicamente activos condujo a las reglas de Sidgwick-Powell en 1940[51], y las ideas adicionales hechas por Gillespie y Nyholm con respecto a los roles estereoquímicos relativos de los enlaces-par y pares solitarios en las moléculas llevaron a su cambio de marca como la teoría de la valencia de la repulsión del par de electrones (VSEPR). Estas relaciones isoelectrónicas pueden interpretarse utilizando el modelo de enlace de valencia, en el que los enlaces-par y los pares solitarios ocupan orbitales hibridados en el átomo central. Dentro del marco orbital molecular, las capas cerradas son una consecuencia del modelo deslocalizado de la misma manera que se encuentra en la descripción de átomos de Schrödinger. Además, los diagramas de Walsh rastrearon la evolución de los orbitales moleculares a medida que las geometrías se alteraron[52] y utilizaron la regla de no cruce para describir la mezcla de MO con las mismas propiedades de simetría. De hecho, el relleno aufbau de los orbitales moleculares forma una parte esencial de la metodología de los orbitales moleculares, y los diagramas de Walsh hicieron mucho para educar a los químicos sobre la forma en que las energías de los orbitales moleculares deslocalizados varían a medida que cambia la geometría molecular.
1.8.6 Limitaciones topológicas de las representaciones de Lewis
La notación de Lewis mediante la cual un enlace químico de dos centros y dos electrones está representado por una línea fue extendida por químicos orgánicos a moléculas con anillos, y la notación de flecha rizada explicaba con éxito los patrones de sustitución de compuestos aromáticos. Estos desarrollos fueron apoyados teóricamente por el uso de Pauling del concepto de resonancia que se desarrolló a partir del modelo de enlace de valencia. Por ejemplo, la resonancia entre las dos formas canónicas alternativas en la figura 13 podría explicar parcialmente la estabilidad y las propiedades aromáticas del benceno y las moléculas relacionadas. Sin embargo, ya en 1931, Hückel estableció que los polienos conjugados cíclicamente[53], como los que se muestran en la figura 13, eran más estables si había 4n+2 electrones involucrados. El primer ejemplo es, por supuesto, el benceno donde n=1 y 4n+2 = 6, es decir, tiene un sexteto aromático. Sin embargo, el ciclobutadieno y el ciclo-octatetraeno son antiaromáticos porque tienen electrones 4n. Esto destacó las limitaciones de basar los argumentos en formas de resonancia como las que se muestran en la parte superior de la Fig. 13. Superficialmente, las formas de resonancia de C4H4 y C8H8 parecerían dar como resultado la estabilización de resonancia, pero las formas de resonancia mostradas no conducen a la deslocalización cíclica. El enfoque de Hückel se vindicó aún más por el aislamiento de sales del anión ciclopentadienilo y el catión tropylium que también tenía electrones de valencia de 6π. El aislamiento de compuestos sándwich de metal de transición del anión ciclopentadienilo, como el ferroceno, que sufre las reacciones de sustitución electrofílica características, y el dianión cicloctatetraeno como el uranoceno subrayó las limitaciones del uso ingenuo de los argumentos de resonancia[54]. Pauling señaló en la Tercera Edición de Nature of the Chemical Bond (1960) que se requerían cientos de estructuras de resonancia para describir adecuadamente la estructura. Estos ejemplos enfatizan los peligros de vincular necesariamente las estructuras de Lewis con las características topológicas de las moléculas.
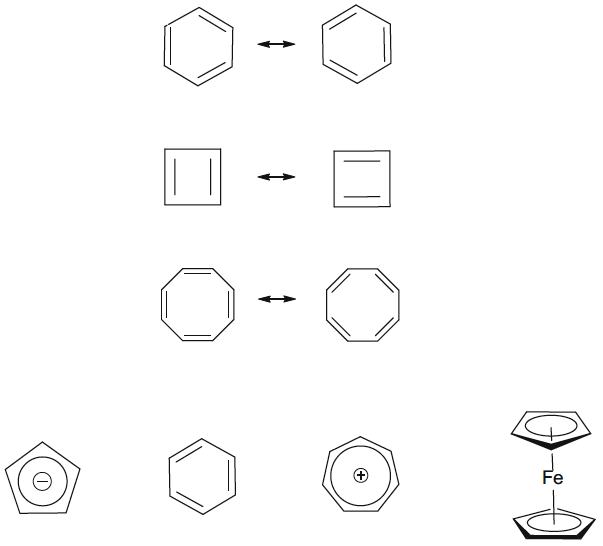
Fig. 13 Formas de resonancia alternativas para benceno (aromático), ciclobutadieno y cicloctatetraeno (antiaromático).
Otros hidrocarburos cíclicos aromáticos y ferroceno se ilustran en la parte inferior
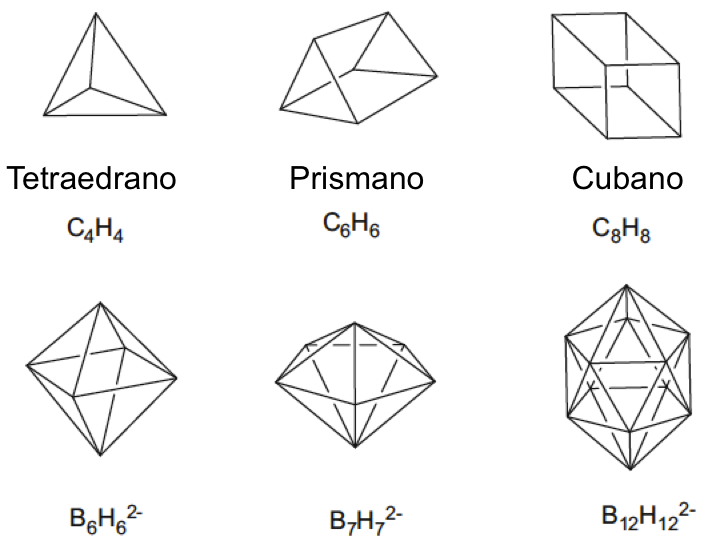
Fig. 14 Ejemplos de moléculas del grupo principal poliédrico. La serie de hidrocarburos que se muestra en la parte superior retiene una conexión entre el número de enlaces de dos electrones y dos centros de C–C y los bordes del poliedro. Esta conexión se pierde para los aniones borano deltaédricos que se muestran en la línea inferior.
Cuando hay una deslocalización extensa, es más apropiado usar modelos orbitales moleculares basados ??en electrones libres.
Una historia similar surgió del estudio de moléculas que tienen estructuras tridimensionales donde se produce una deslocalización extensa. La descripción de Lewis proporciona una buena descripción de anillos orgánicos y tres moléculas de CnHn poliédricas conectadas que se ilustran en la Fig. 14. Los ángulos pequeños en esas moléculas con caras triangulares y cuadradas requerían químicos orgánicos para considerar las consecuencias de los enlaces C-C tensos o doblados, pero esencialmente se mantiene en la imagen de Lewis. Sin embargo, la conexión topológica entre el número de bordes del poliedro y el número de enlaces de dos electrones y dos centros de Lewis ya no es válido para boranos deltaédricos. Longuet–Higgins fue el primero en desarrollar una analogía tridimensional de la aproximación Hückel que explicaba satisfactoriamente el enlace en los aniones borano BnHn^2- y los aniones boruro B62- y B122-. Estos deltaedros esféricos se caracterizan por la unión de n+1 de los orbitales moleculares esqueléticos, que no tienen conexión geométrica directa con el número de caras (2n-4) o bordes (3n-6) de deltaedro. Posteriormente, este enfoque se extendió a los aniones borano deltaédricos y formó la base de la teoría del par de electrones esqueléticos poliédricos (PSEPT), que proporcionó un análogo tridimensional de la teoría VSEPR y una extensión a tres dimensiones de la teoría de la deslocalización cíclica bidimensional de Hückel. Wade y Mingos en la década de 1970 fueron los principales contribuyentes a estos desarrollos[55]. La conexión con el enfoque Hückel fue respaldada por Stone, quien desarrolló un modelo armónico esférico de electrones libres para el grupo principal deltaédrico y los grupos de metales de transición en la década de 1980.
1.8.7 Analogías isolobal
La aparición de compuestos organometálicos de grupo principal isoestructural y de metal de transición que se ajustan al octeto o las reglas de 18 electrones sugiere posibles conexiones electrónicas entre las dos clases de compuestos. Los compuestos del grupo principal ilustrados en la figura 14 se caracterizan por los siguientes recuentos de electrones de valencia total: tres electrones 5n conectados (por ejemplo, CnHn) y electrones deltaédricos 4n+2.
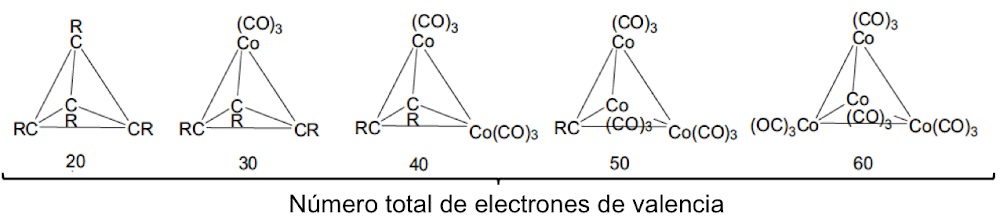
Fig. 15 Ejemplos de grupos iso-estructurales, donde el número total de electrones de valencia aumenta en 10 electrones por cada átomo de metal de transición introducido.
(por ejemplo, BnHn2). Los carbonilos de metales de transición forman compuestos análogos con electrones de valencia 15n para moléculas poliédricas de tres conexiones, p.ej. tetraédrica-[Rh4(CO)12] y 14n+2 para moléculas poliédricas deltaédricas, p.ej. octaédrica [Co6(CO)14]4-. Estas relaciones sugieren una serie de compuestos isoestructurales cuyo recuento total de electrones de valencia aumenta en 10 electrones por cada fragmento de metal de transición introducido en el esqueleto poliédrico del grupo principal. La Figura 15 proporciona ejemplos específicos de grupos tetraédricos que se ajustan a esta generalización.
Esta conexión entre el grupo principal y los grupos de metales de transición se ha basado en la analogía isolobal, que relaciona las propiedades de los fragmentos constituyentes con las simetrías y energías de los orbitales fronterizos[56]. Curiosamente, la ubicación de los puentes terminales ligandos de carbonilo no tiene que definirse para que la relación funcione. De hecho, se demostró en una etapa temprana que la analogía isolobal depende principalmente de la similitud de los orbitales moleculares antienvejecimiento, que permanecen desocupados en ambas series de moléculas.
1.8.8 Núcleo y electrones de valencia
La descripción mecánica cuántica del átomo que resultó de la descripción mecánica de la onda de Schrödinger del átomo de hidrógeno condujo a una comprensión mucho más profunda de los factores electrónicos que fueron responsables de la clasificación periódica de Mendeleev y la descripción de Lewis y Kossel del enlace químico y la valencia[57]. Dado que el enfoque de Lewis hace una distinción clara entre los electrones de núcleo y de valencia, era importante establecer si la descripción mecánica cuántica y las mediciones espectroscópicas asociadas respaldaban esta suposición. Cuando el modelo de Schrödinger se extendió al átomo de polielectrón, se supuso que las partes angulares de la solución eran las mismas que las desarrolladas para el átomo de hidrógeno, pero la parte radial se modificó para tener en cuenta las diferencias en carga nuclear y efectos de repulsión de electrones. Estos efectos eliminan las degeneraciones de los orbitales de los átomos de hidrógeno con el mismo número cuántico principal n pero diferentes números cuánticos, es decir, las energías de ns–np–nd, etc., ya no son iguales. Los estudios espectrales sobre transiciones electrónicas utilizaron las consecuencias de simetría del modelo para confirmar el orden de los niveles de energía en los átomos y proporcionar evidencia empírica con respecto a la definición de electrones centrales y de valencia en los átomos. Estos resultados experimentales fueron respaldados por cálculos basados ??en mecánica cuántica. Los estudios teóricos proporcionaron información sobre las probabilidades de la distribución de electrones en los orbitales atómicos y condujeron a la definición de rrmax, el radio orbital más probable. El desarrollo de una cristalografía de rayos X confiable condujo a datos sobre distancias interatómicas en metales, sales y compuestos moleculares. Por ejemplo, en un metal cristalino, el radio metálico de un átomo metálico puede definirse como 1⁄2 la distancia internuclear entre átomos metálicos vecinos en estado sólido. Esta distancia internuclear se correlaciona bien con la rmax calculada de los orbitales de valencia de metales como se muestra en la figura 16. Por lo tanto, no es irrazonable que la superposición máxima entre orbitales de átomos metálicos adyacentes ocurra cuando los átomos metálicos están separados por una distancia cercana a 2rrmax para los orbitales de valencia ns. Los electrones centrales no contribuyen significativamente a la unión metal-metal porque sus radios orbitales son mucho más pequeños, como se muestra en la Tabla 5. Los núcleos tienen volúmenes entre uno y dos órdenes de magnitud más pequeños que los de los electrones de valencia. Los orbitales de valencia np vacíos de los metales alcalinos son capaces de superponerse con los orbitales ns y np de los átomos adyacentes, pero su contribución no se manifiesta en las longitudes de enlace observadas.
Fig. 16Gráfico de rmax y radio metálico para metales alcalinos
Tabla 5 Valores de rmax (en relación con H rmax = 52.918 pm) para los átomos de metales alcalinos. Los números en negrita se refieren a los orbitales de valencia
Para los elementos que se encuentran hacia el centro de una fila de la tabla periódica, los orbitales de valencia ns y np contribuyen significativamente al enlace, y las gráficas de ns y np rmax contra los radios covalentes observados sugieren que el enlace está cada vez más dominado por los orbitales np (Fig. 17) para los elementos más pesados ??del grupo. Para el carbono, tanto 2s como 2p contribuyen significativamente al enlace covalente, pero el carácter ns en los enlaces disminuye para los elementos más pesados. Esto se refleja en los ángulos de enlace en las moléculas XYn cuando n = 2 o 3.
Fig. 17 Gráficos de rmax y radios covalentes determinados estructuralmente para los elementos del grupo 14 C, Si, Ge, Sn y Pb
Estudios teóricos recientes y determinaciones experimentales precisas de las densidades de electrones en las moléculas han confirmado que la mayoría de los electrones sí forman un núcleo concentrado cerca del núcleo que parece muy atómico. La densidad electrónica es muy monotónica a medida que aumenta la distancia radial desde el núcleo. Los químicos a veces consideran erróneamente que la densidad de electrones en los núcleos de los átomos tiene máximos externos correspondientes a las estructuras de la cubierta. Los núcleos de electrones internos son entidades casi transferibles y, en consecuencia, respaldan la partición de cuidado de valencia propuesta por Lewis y Kossel, y esta propiedad se utiliza en aproximaciones de núcleos congelados.
Un pseudo potencial es un potencial que reemplaza efectivamente el potencial atómico de todos los electrones, de tal manera que se eliminan los estados centrales y los electrones de valencia se describen mediante funciones de pseudo ondas con significativamente menos nodos. Solo los electrones de valencia químicamente significativos se tratan explícitamente, mientras que los electrones centrales están “congelados”. Ellos y los núcleos se tratan como núcleos de iones rígidos no polarizables. Esta restricción de núcleo congelado se puede relajar actualizando de forma coherente el pseudo potencial con el entorno químico en el que está incrustado. Los pseudo potenciales derivados de los primeros principios se basan en un estado de referencia atómico, que requiere que el pseudo y todos los electrones de valencia electrónica de estados propios tienen las mismas energías y densidad fuera de un radio de corte de núcleo elegido.
Aunque Lewis asumió que todos los electrones de valencia en la capa externa de un átomo tenían la misma capacidad de enlace, esta visión tuvo que modificarse a la luz del conocimiento teórico y empírico posterior. Específicamente, las capacidades de enlace de los electrones de valencia dependen no solo del número cuántico principal sino también de los orbitales de valencia específicos. La Tabla 6 resume la forma en que la hibridación de los orbitales en compuestos orgánicos que tienen enlaces simples C-C está influenciada por la hibridación de los átomos de carbono. Las longitudes de los enlaces disminuyen a medida que aumenta la cantidad de caracteres s.
Las diferencias resultantes de las variaciones en las funciones de distribución radial de diferentes orbitales de valencia se vuelven más pronunciadas para los átomos que tienen orbitales de valencia nd y nf, porque están significativamente más contraídos que los orbitales (n+1)s y (n+1)p y por lo tanto, comportarse de una manera más básica.
Tabla 6 Influencia de la hibridación sp^x en las longitudes de enlace C–C
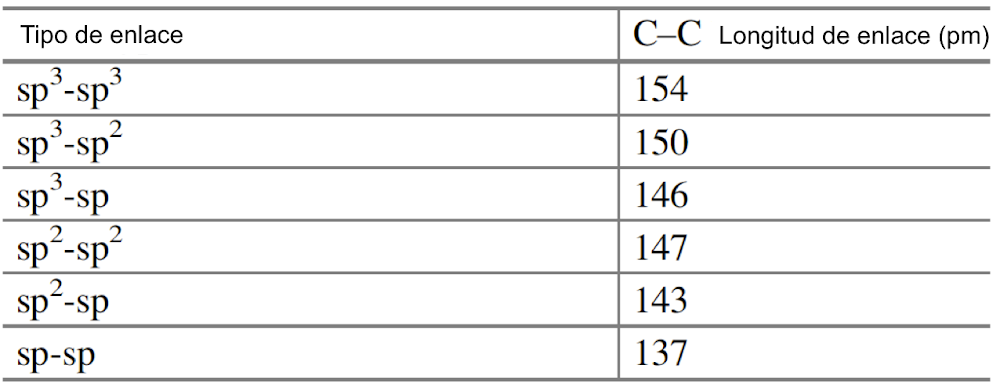
Los metales de transición tienen orbitales de valencia nd, (n+1)s y (n+1)p, pero sus funciones de distribución radial son muy diferentes. Para el cromo, el máximo de la función de distribución radial del orbital 3d es 46pm, mientras que para el orbital 4s, es 161, es decir, más de tres veces mayor. La naturaleza mucho más contraída de nd en relación con (n+1)s y (n+1)p significa que son mucho más centrales. En consecuencia, los compuestos de los metales de transición muestran una dualidad de propiedades. Para ligandos más grandes que no se superponen fuertemente con los orbitales nd, se comportan como si los orbitales nd fueran similares a núcleos y formaran series de compuestos que tienen estructuras similares. Estos compuestos se han descrito como compuestos isoestequiométricos. Los ejemplos de haluros metálicos que son isoestequiométricos se resumen en las Tablas 7, 8, 9 y 10. En estas tablas, las garrapatas representan compuestos conocidos y cruzan compuestos desconocidos. Los ligandos que tienen radios más pequeños o forman enlaces múltiples fuertes con el metal son capaces de superponerse fuertemente con los orbitales nd y formar enlaces covalentes fuertes. Dichos compuestos obedecen la regla EAN, y algunos ejemplos específicos se resumen en la Tabla 10.
En consecuencia, los carbonilos de metales de transición se comportan como compuestos del grupo principal donde la estequiometría cambia para satisfacer la regla del gas inerte, y esta similitud sustenta las relaciones isolobales. Los orbitales f de los lantánidos están aún más contraídos y, en consecuencia, son más centrales. En resumen, la distinción entre los electrones de valencia y núcleo inicialmente hechos por Lewis y Kossel puede justificarse y es generalmente aplicable a los elementos representativos, pero tiene sus limitaciones cuando se aplica a elementos con orbitales d y f parcialmente llenos, y los metales de transición muestran características Jekkyl y Hyde que dependen de la capacidad de los ligandos para superponerse con los orbitales d.
Tabla 7 Dihaluros de metales divalentes isoestequiométricos
Tabla 8 Haluros metálicos trivalentes isoestequiométricos
Tabla 9Complejos metálicos isoestequiométricos
Tabla 10 Ejemplos de complejos metálicos que se ajustan a la regla EAN
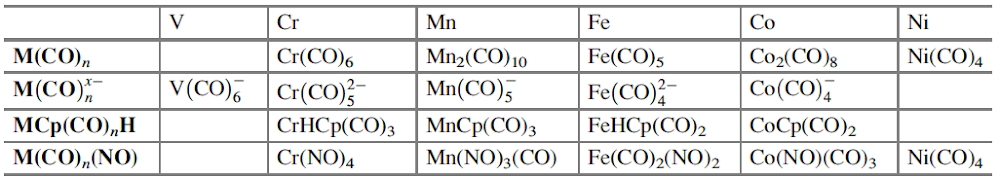
1.9 Molécula de electrones impares
Lewis señaló en su artículo de 1913 que la gran mayoría de las moléculas tienen un número par de electrones, y esto formó una observación fundamental en el desarrollo del concepto del enlace par de electrones. Señaló que el NO y el NO2 proporcionaban ejemplos raros de excepciones y que los efectos estéricos eran importantes para influir en la posibilidad de aislar moléculas de electrones extraños. En los últimos años, la incorporación de ligandos estéricamente exigentes ha dado como resultado una amplia gama de moléculas que no confirman la descripción de Lewis o son paramagnéticas[58]. Además, el estudio de moléculas en fase gaseosa y en condiciones de aislamiento de matriz ha dado como resultado la caracterización estructural de moléculas que no son necesariamente las moléculas más estables en condiciones estándar. También los métodos espectroscópicos han resultado en el estudio de moléculas en sus estados excitados, y las geometrías establecidas por estos estudios han proporcionado desafíos adicionales para el modelo de Lewis-Kossel, porque las moléculas tienen métricas diferentes y, a veces, geometrías en sus estados excitados.
Los metales de transición, los lantánidos y los actínidos proporcionan muchos ejemplos de compuestos con electrones no apareados debido al carácter de núcleo parcial de las subcapas nd y nf para estos grupos de elementos, como se discutió anteriormente.
1.10 Descripción mecánica cuántica del enlace químico
Gavroglu y Simoes escribieron una descripción histórica detallada del desarrollo temprano de la química cuántica. Burrau publicó la primera descripción cuántica matemáticamente completa de las especies de enlace químico más simples, es decir, el ion molecular de hidrógeno, H2+, en 1927[59]. Esto demostró que una descripción mecánica cuántica del enlace químico era viable, pero los métodos matemáticos utilizados no podían extenderse fácilmente a las moléculas que contienen dos o más electrones. Heitler y London propusieron un enfoque más flexible, pero menos exacto, en el mismo año, y esto formó la base de lo que llegó a describirse como la teoría del enlace de valencia[60]. Reconoció las importantes contribuciones hechas por Pauli. El principio de exclusión de Pauli establece que dos partículas con espín de medio entero no pueden ocupar el mismo estado cuántico simultáneamente[61]. Para los electrones en átomos y moléculas, es imposible que dos electrones tengan los mismos valores de los cuatro números cuánticos (n, l, ml y ms). Para dos electrones que residen en el mismo orbital, n, l y ml son iguales, por lo que ms debe ser diferente y los electrones tienen espines opuestos. La tendencia de las moléculas a tener pares de electrones se debe al principio de Pauli que permite un máximo de dos electrones en la misma región del espacio. Se puede formar un enlace químico con un solo electrón como en H2+. Por lo tanto, el enlace químico no se debe a la formación de un par de electrones como lo sugiere Lewis, sino a la superposición de las funciones de onda. Al mismo tiempo, Hund, Urey y Lennard-Jones, quienes también sugirieron métodos para derivar las estructuras electrónicas de las moléculas F2 (flúor) y O2 (oxígeno), desarrollaron la combinación lineal de aproximación del método de orbitales moleculares (LCAO), a partir de principios cuánticos básicos[62]. Todas estas descripciones aproximadas de enlaces químicos se basaron en la descripción mecánica cuántica de Schrödinger de los orbitales atómicos que se había desarrollado exactamente para el átomo de hidrógeno y posteriormente se extendió de manera aproximada a los átomos de polielectrones. Las ecuaciones para enlaces de electrones en átomos multielectrónicos no pudieron resolverse analíticamente, pero las aproximaciones dieron muchas predicciones y resultados cualitativos, que podrían ser utilizados por los químicos.
En 1933, James y Coolidge convencieron a la comunidad científica de que la teoría cuántica[63] podía dar resultados de acuerdo con el experimento de moléculas simples como el H2. A diferencia de todos los cálculos anteriores, que usaban funciones que dependían solo de la distancia del electrón desde los núcleos atómicos, usaban funciones que también agregaban explícitamente la distancia entre los dos electrones. Con hasta 13 parámetros ajustables, obtuvieron un resultado muy cercano al resultado experimental para la energía de disociación. Las extensiones posteriores utilizaron hasta 54 parámetros y dieron un excelente acuerdo con los experimentos. Sin embargo, este enfoque tuvo en cuenta cuantitativamente la energía y la métrica de los enlaces químicos a expensas de disminuir los rasgos característicos de los modelos de enlace de valencia y modelos orbitales moleculares.
1.10.1 Modelo del enlace de valencia
Según el modelo de Heitler-London, se forma un enlace covalente por la superposición de orbitales atómicos de valencia medio llenos de dos átomos, cada uno con un electrón no apareado. Sus cálculos para dos átomos de hidrógeno neutros, que tuvieron en cuenta la intercambiabilidad de los dos electrones a medida que los átomos se acercaban entre sí, mostraron que una situación más estable (enlace) resultó solo cuando los espines de los electrones eran antiparalelos. El modelo de pares de electrones de Lewis, por lo tanto, requería no solo un par de electrones sino también que tenían espines opuestos. Por lo tanto, la unión covalente se representó como un efecto mecánico puramente cuántico ya que el espín electrónico no tenía un análogo clásico. Sus cálculos dieron una explicación razonable de la energía de disociación y la distancia internuclear en la molécula de hidrógeno. La superposición de las funciones de onda de los dos átomos se asoció con un aumento en la densidad de electrones en la región de enlace cuando los electrones son antiparalelos. Para compuestos más complejos, la teoría se centra en pares de electrones dentro de las moléculas (la aproximación de emparejamiento perfecto) y sugiere que estos son dominantes en la formación de enlaces. Cuando varias estructuras perfectamente emparejadas tienen energías similares, se permite que la molécula resuene entre ellas y se reduce la energía total. Matemáticamente, la resonancia se representa agregando funciones de onda adicionales. Por lo tanto, una estructura de enlace de valencia incorporó el emparejamiento de electrones central al enlace covalente de Lewis, pero si no se puede escribir una estructura de Lewis, porque no representa adecuadamente la simetría o polaridad de la molécula, entonces se utilizan varias funciones de onda de enlace de valencia.
Pauling desarrolló el modelo de Heitler-Londres enfatizando la importancia del enlace electrónico-químico compartido y enfatizó que la valencia química era el resultado del principio de exclusión de Pauli y el fenómeno de resonancia mecánica cuántica. Señaló “No me considero un extraño que traiga algo del exterior, sino un miembro de un grupo que lleva a cabo el trabajo iniciado por el profesor Lewis en 1916, desde que me enteré del enlace del par de electrones, en 1920, me he dedicado mis esfuerzos por intentar comprender las propiedades de las sustancias desde este punto de vista”. Pauling y Slater publicaron casi simultáneamente artículos que tenían muchas características comunes, pero Pauling fue más claro en su deseo de proponer una solución universal[64]. “Las ecuaciones mecánicas cuánticas permiten la formulación de un amplio y poderoso conjunto de reglas para el enlace del par de electrones que complementa las de Lewis. Estas reglas proporcionan información sobre las fuerzas relativas de los enlaces formados por diferentes átomos, los ángulos entre los enlaces, la rotación libre o la falta de rotación libre sobre los ejes de enlace, la relación entre los números cuánticos de los electrones de enlace y el número y la disposición espacial de los enlaces”. Pauling utilizó las partes angulares de la función de onda de Schrödinger de un átomo para proporcionar una simplificación que produjo muchas nuevas ideas importantes sobre las propiedades y fuerzas de los enlaces. La claridad de presentación de Pauling y su conocimiento enciclopédico de los hechos químicos proporcionaron un papel importante para cerrar la brecha entre la química y la nueva teoría cuántica desarrollada por los físicos.
Pauling demostró que la fuerza de enlace de un enlace covalente era mayor si dos orbitales atómicos se dirigen de modo que la densidad máxima de electrones se localice entre los núcleos. Utilizó las partes angulares de las soluciones de la ecuación de Schrödinger para desarrollar orbitales hibridados sp3, sp2 y sp que representaban las geometrías de las moléculas orgánicas y del grupo principal y los orbitales híbridos d2sp3 y dsp2 para complejos de metales de transición. Estos orbitales híbridos se construyen utilizando las relaciones de ortagonalidad, que resultan de las ecuaciones de onda para las partes angulares de las soluciones de Schrödinger. Surgen naturalmente de la descripción de la onda mecánica cuántica, que se expresa como funciones armónicas esféricas. Pauling también extendió el concepto de resonancia para explicar la deslocalización de electrones en sistemas π conjugados y la polaridad de los enlaces. Esto requirió la introducción de una escala de electronegatividad para describir las capacidades relativas de los átomos para atraer electrones.
La Naturaleza del Enlace Químico de Pauling publicada en 1938 tuvo un inmenso impacto en el pensamiento químico[65]. Introdujo a los químicos sobre la importancia del cultivo cuántico desarrollando los conceptos de resonancia y subrayando la importancia de las superposiciones orbitales atómicas y la hibridación para influir en las fuerzas y geometrías de las moléculas. También desarrolló principios empíricos para subrayar la transferibilidad de las longitudes y energías de enlace y la importancia de la electronegatividad en la definición del carácter iónico parcial de los enlaces. Extendió el modelo de enlace iónico de Kossel y articuló un conjunto de principios para comprender las estructuras de los sólidos infinitos y sus métricas importantes. Subrayó la importancia de los enlaces de hidrógeno no solo en estos compuestos sino también en los principios estructurales, subyacentes al plegamiento de proteínas y enzimas.
La popularidad temprana de los métodos de enlace de valencia disminuyó, porque los cálculos de los orbitales moleculares eran más susceptibles de escribir algoritmos informáticos eficientes. Recientemente, la programación de los métodos de enlace de valencia ha mejorado porque también se benefició del rápido desarrollo de la tecnología informática y se han desarrollado programas que son competitivos en precisión y economía con los programas para el método Hartree-Fock y otros métodos basados ??en orbitales moleculares. Estos desarrollos se deben y han sido descritos por Gerratt, Cooper, Karadakov y Raimondi y resumidos por Li y McWeeny en 2002, van Lenthe y compañeros de trabajo en 2005 y las revisiones de Shaik e Hiberty en 2008[66].
En su forma más simple, los orbitales atómicos superpuestos se reemplazan por orbitales que se expanden como combinaciones lineales de las funciones básicas basadas en átomos, formando combinaciones lineales de orbitales atómicos (LCAO). Esta expansión está optimizada para proporcionar la energía más baja. La teoría moderna del enlace de valencia es, por lo tanto, una extensión del método de Coulson-Fischer. Este procedimiento proporciona buenas energías sin incluir estructuras iónicas. La teoría moderna del enlace de valencia reemplaza la combinación lineal simple de los dos orbitales atómicos con una combinación lineal de todos los orbitales en un conjunto de bases más amplio. Los dos orbitales de enlace de valencia resultantes parecen un orbital atómico en un átomo de hidrógeno ligeramente distorsionado hacia el otro átomo de hidrógeno.
El método de enlace de valencia generalizado (GVB), desarrollado por Goddard en 1970, es uno de los métodos de enlace de valencia más simples y antiguos que utilizan orbitales flexibles de manera general. La teoría generalizada de Coulson-Fischer para la molécula de hidrógeno mencionada anteriormente se utiliza para describir cada par de electrones en una molécula[67]. Los orbitales para cada par de electrones se expanden en términos del conjunto de bases completo y no son ortogonales. Los orbitales de diferentes pares se ven obligados a ser ortogonales. Esta condición simplifica los cálculos, pero puede dar lugar a algunas dificultades[68].
1.10.2 Teoría del orbital molecular
El interés de Mulliken en los niveles electrónicos en las moléculas en la década de 1920 fue estimulado por las sugerencias de que sus espectros moleculares tenían similitudes con los espectros atómicos y se podían discernir relaciones definidas para las moléculas isostéricas. Descubrió que la analogía espectroscópica entre las moléculas isostéricas podría extenderse a átomos con la misma cantidad de electrones, y esta relación conduciría posteriormente al enfoque de átomo unido. Él y Birge clasificaron los estados electrónicos en moléculas diatómicas utilizando la misma clasificación de Russell-Saunders utilizada anteriormente para los estados atómicos. El análisis teórico de Hund de la naturaleza de los estados electrónicos en las moléculas, por lo tanto, resultó oportuno para Mulliken y lo llevó a publicar un resumen de la teoría y proporcionar pruebas experimentales adicionales que la respaldan. En la teoría de los orbitales moleculares, Hund mostró cómo el concepto de los orbitales atómicos y los procedimientos matemáticos desarrollados para definirlos podrían extenderse a las moléculas.
Sin embargo, en una molécula, los electrones experimentan los campos ejercidos por todos los núcleos y los otros electrones. Los orbitales moleculares resultantes, que en la aproximación más simple se describen como combinaciones lineales de los orbitales atómicos de cada centro, se deslocalizan sobre la molécula completa. Cada orbital molecular está definido por su energía, y sus propiedades de simetría están definidas por el grupo de puntos de la molécula. Las simetrías podrían estar relacionadas con las de los átomos esféricos utilizando el descenso de los procedimientos de simetría desarrollados a partir de la teoría de grupos. En una aproximación de un electrón, las energías de los orbitales moleculares están determinadas por sus características nodales y su distribución sobre las moléculas. En manifestaciones más sofisticadas, se introducen los efectos de repulsión y correlación entre electrones. La superposición entre los orbitales y la diferencia de electronegatividad entre los átomos, por lo tanto, influye en la distribución de la densidad electrónica. El espectro resultante de los orbitales moleculares está ocupado por el número total de electrones en la molécula utilizando el procedimiento de aufbau, y las funciones de onda obedecen al principio de exclusión de Pauli. Los orbitales moleculares más estables tienen pocos nodos radiales y angulares y se describen como orbitales moleculares de enlace, y a medida que se introducen más nodos, los orbitales se vuelven progresivamente más anti-enlace. En las moléculas diatómicas simples, la separación de energía entre los enlaces orbitales moleculares de y antienlace está influenciada principalmente por la superposición entre los orbitales atómicos constituyentes y las electronegatividades de los átomos. Entre estas dos clases de orbitales moleculares, se observan orbitales moleculares que no se unen y que generalmente tienen ganglios que se encuentran en posiciones que anulan las superposiciones orbitales del vecino. Se realiza una conexión aproximada con la descripción del enlace de Lewis tomando la diferencia en el número de orbitales moleculares de enlace y antienlace doblemente ocupados. Esto a menudo coincide con el número de enlaces de pares de electrones en la descripción de Lewis. La ocupación de orbitales moleculares sin unión tiene un efecto neutral y, por lo tanto, la ocupación de estos MO sin enlace puede conducir a una serie de moléculas e iones relacionados con el mismo orden de unión formal, por ejemplo el catión alilo, el radical y el anión tienen el mismo orden formal de enlace C–C porque los electrones de adición ocupan un orbital molecular π no enlazante localizado en los átomos de carbono externos. Mulliken reconoció la contribución de Lewis de la siguiente manera:
La mejor teoría química de la valencia que cubre todos los tipos de compuestos generalmente se acepta como la desarrollada por G.N. Lewis. En gran medida, las características esenciales de esta teoría siguen vigentes, aunque su significado se ha hecho más claro y más específico al interpretarlas a la luz de la teoría cuántica.
Él, de manera un tanto traviesa, hizo el siguiente comentario sobre la relación entre su análisis orbital molecular y el modelo de pares de electrones de Lewis: “Ahora tengo un argumento favorito de que el enlace de pares de electrones de Lewis se describe mejor por un par de electrones en un orbital molecular que por el vínculo Heitler-Londres. Si el enlace químico tiene alguna polaridad, es necesario agregar un término iónico, que es un Heitler-London más un término iónico, para representar el enlace. Esa es una descripción bastante desordenada, mientras que el orbital molecular no es el espectroscópico sino el orbital molecular químico, el orbital molecular deslocalizado encaja muy bien con el concepto de Lewis”.
Reservó sus principales críticas para el enfoque de Heitler–Londres, Pauling–Slater (HLPS):
Las teorías de HLPS podrían llamarse teorías de emparejamiento de electrones si Lewis le llama una teoría de pares de electrones. También debe señalarse que el par de electrones HLPS difiere considerablemente de la concepción de Lewis del enlace par de electrones, en que los electrones están mucho menos asociados; a este respecto, se acerca mucho más a la verdad que la concepción de Lewis: Pauling y Slater consideran que un doble enlace son simplemente dos enlaces simples que sobresalen de cada átomo en diferentes direcciones, y tratan el triple enlace de manera similar. De esta manera, no están muy de acuerdo con Lewis, ni con los resultados obtenidos de la teoría de los orbitales moleculares.
Dado su origen cercano con el análisis espectral, la descripción orbital molecular se usó más ampliamente en los primeros días para describir las estructuras electrónicas de las moléculas en sus estados excitados, es decir, moléculas en las que uno o más electrones han sido promovidos desde el enlace y no enlace del estado fundamental de orbitales moleculares a antienlazamiento de orbitales moleculares. En estos estados excitados, las longitudes de enlace cambian para reflejar las diferentes ocupaciones de los orbitales moleculares de enlace y antienlace, y también para las moléculas poliatómicas, la geometría del estado excitado puede diferir de la del estado fundamental[69].
Mulliken confirmó los diagramas orbitales moleculares iniciales para las moléculas poliatómicas utilizando datos espectrales moleculares y posteriormente se verificaron mediante cálculos orbitales moleculares de campo autoconsistentes. Mulliken también desarrolló el enfoque del diagrama de correlación que utilizaba argumentos de simetría y la regla de no cruce. Dichos diagramas muestran cómo cambian las energías orbitales moleculares en función de las distancias internucleares, R. Cuando R es muy grande, los orbitales moleculares deben ser los mismos que los de los átomos aislados y progresar a través de los orbitales moleculares para la molécula poliatómica con las apropiadas distancias internucleares Requil, y como R tiende a cero, los orbitales moleculares deben transformarse en los orbitales atómicos del átomo unido. La correlación de esos orbitales moleculares con las mismas características de simetría conduce al diagrama de correlación. Estos diagramas de correlación deben cumplir con la regla de no cruce que establece que los orbitales atómicos y moleculares con las mismas simetrías no pueden cruzarse. Este tipo de análisis proporciona un marco teórico para comprender por qué la regla del gas inerte es aplicable tanto a las moléculas como a los átomos[70]. La siguiente serie de moléculas ilustra las relaciones isoelectrónicas:
8 electrones de valencia
Posteriormente, estos diagramas de correlación orbital desempeñaron un papel muy importante en el desarrollo de los diagramas de Walsh y, lo que es más importante, en la aclaración de las reglas de simetría orbital desarrolladas por Woodward y Hoffmann, que explicaron las estereoquímicas de las reacciones pericíclicas de las moléculas orgánicas[71].
El desarrollo de la teoría de la órbita molecular en la década de 1950 siguió dos caminos distintos. En ese momento, las computadoras digitales (y de hecho las calculadoras electrónicas) no estaban ampliamente disponibles y los cálculos cuantitativos consumían mucho tiempo, y los desarrollos conceptuales en química orgánica se basaron en gran medida en la aproximación de Hückel. Sin embargo, Longuet–Higgins y Dewar fueron capaces de proporcionar un fuerte modelo conceptual basado en orbitales moleculares para tratar el enlace en sistemas orgánicos π, que utilizaban las propiedades alternativas de estas moléculas. Su aplicación de las ideas de la teoría de la perturbación resultó en una metodología aproximada y en gran parte pictórica. Dewar (no conocido por su subestimación) señaló en su libro “que la teoría general de la química orgánica (basada en la teoría de la perturbación) no era más difícil de aplicar que la teoría de la resonancia, sino infinitamente más poderosa y versátil[72]”. Fukui desarrolló argumentos similares de la teoría de la perturbación, y sus estudios enfatizaron el importante papel desempeñado en las reacciones de las moléculas orgánicas por sus orbitales moleculares fronterizos. Las energías y las características nodales del orbital molecular ocupado más alto (HOMO) y el orbital molecular desocupado más bajo (LUMO) debían ser particularmente importantes en el análisis de las reacciones orgánicas, y estos acrónimos se han convertido en parte del lenguaje del químico moderno. Hoffmann desarrolló simultáneamente la metodología extendida de Hückel para los enlaces σ de moléculas orgánicas y proporcionó una plataforma para confirmar las ideas de simetría orbital desarrolladas con Woodward[73]. Hoffmann y Fukui compartieron el Premio Nobel por sus contribuciones. Posteriormente, Pearson[74] mostró que la notación de enlace de Lewis podría incorporarse a las reglas de Woodward Hoffmann teniendo en cuenta las simetrías de las combinaciones lineales de los orbitales fronterizos. Desde entonces, Hoffmann ha utilizado la metodología extendida Hückel con mucho éxito para analizar una amplia gama de problemas de valencia en coordinación, química organometálica y de estado sólido[75].
1.10.3 Modelos de enlaces sinérgicos
La propuesta de que el enlace en los pares ácido-base de Lewis puede describirse como un enlace covalente polarizado se reconoció poco después de la publicación de los documentos de Lewis en 1916, y Sidgwick desempeñó un papel importante al proporcionar una notación adecuada y aplicarla a la coordinación de compuestos de Werner. Los carbonilos de metales de transición, por ejemplo Ni(CO)4, Fe(CO)5 y Cr(CO)6, cumplieron con la regla EAN, pero proporcionaron algunas dificultades para el principio de electroneutralidad. Los estudios de difracción de electrones mostraron que los enlaces M–C eran más cortos que los sugeridos por consideraciones de radios covalentes y las longitudes de enlace C–O solo un poco más largas que las de CO libre. Pauling en la segunda edición de la naturaleza del enlace químico propuso que el enlace “implica un doble enlace de níquel a carbono” (las cursivas son suyas) y propuso que la forma canónica que se muestra en la Fig. 18 hizo una contribución significativa. Añadió “Esta estructura es más satisfactoria que la estructura de enlace único por la siguiente razón: hace que el átomo de níquel y los otros átomos sean neutros, mientras que las estructuras de enlace único colocan una carga negativa cuádruple en el átomo de níquel, es en su comportamiento general electropositivo en lugar de electronegativo”. La sugerencia de Pauling prevaleció a lo largo de la década de 1940, e Hieber, el líder en el campo[76], aceptó la posibilidad de una doble vinculación y sintió que eran consistentes con los datos de momento dipolar. Chatt[77] y Wilkinson[78] adaptaron el modelo de Pauling a los análogos de carbonilos PF3, invocando la participación de los orbitales 3d vacantes en fósforo (Fig. 18). Por lo tanto, la posibilidad de que ciertos ligandos pudieran funcionar como bases de Lewis y ácidos de Lewis ganó vigencia. Sin embargo, la mayoría de los químicos inorgánicos no tenían los antecedentes teóricos para reformular el modelo dentro de un marco orbital molecular, lo que requería una comprensión de las restricciones de simetría que rigen las interacciones orbitales. En abril de 1950, en una conferencia celebrada en Montpelier, Dewar presentó el documento “Una revisión de la teoría del complejo π” y, en respuesta a una pregunta, desarrolló el modelo de enlace sinérgico ilustrado en la Fig. 19. Esto condujo al desarrollo detallado y completo del modelo de enlace sinérgico para complejos metal-alqueno, que se ha convertido en una piedra angular importante de la química organometálica. Dewar expresa los conceptos básicos muy completamente:
Los electrones d en metales pesados, bromo, etc. tienen la simetría correcta para interactuar con el antienlazamiento molecular π de una olefina, en complejos π de la olefina y el átomo pesado. Si este último lleva electrones d, puede formar un segundo enlace molecular dativo con el orbital molecular π antienvejecimiento vacante, en dirección opuesta al enlace molecular normal. Esto se ilustra esquemáticamente a continuación, identificando las fases de los lóbulos de los orbitales para mostrar las propiedades de simetría. El orbital s de Ag+ tiene la simetría incorrecta para interactuar con el antienlace orbital π-molecular. Los dos orbitales moleculares son, por lo tanto, distintos. La combinación de estos dos enlaces dativos dirigidos opuestamente dejaría a la olefina mucho menos cargada de lo que hubiera estado en un complejo π normal; esto explicaría la baja reactividad de los complejos π de las olefinas con metales, donde la energía de enlace de los electrones d es baja, y también las diferencias en la reactividad de los diferentes metales, ya que la estabilidad de los dos enlaces se verá afectada de manera diferente por los cambios en estructura general.
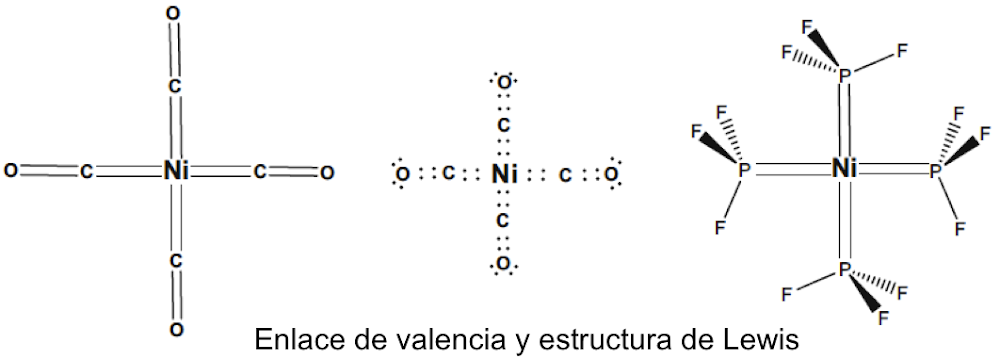
Fig. 18 Estructura de Lewis propuesta por Pauling para Ni(CO)4 y su adaptación a los complejos PF3 relacionados
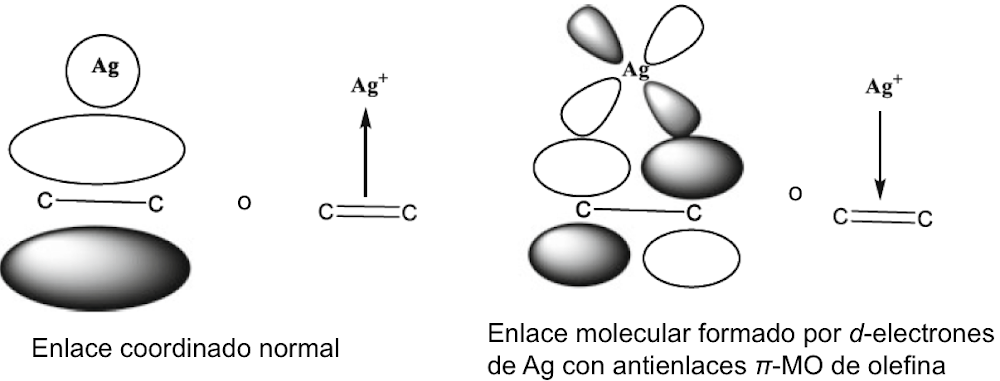
Fig. 19 Ilustración de Dewar del enlace sinérgico en [Ag(C2H4)]+ (sus signos +/- para designar las relaciones de fase han sido reemplazados por sombreado)
Como resultado de las contribuciones de Ingold, los químicos orgánicos estaban muy familiarizados con los reactivos nucleofílicos y electrofílicos, pero Dewar estableció que los ligandos como el CO podrían actuar simultáneamente como nucleófilos y electrófilos (es decir, son ambifílicos) y la interacción sinérgica resultante no solo mejora la fuerza del enlace metal-ligando sino que también fomenta la electroneutralidad. Al enfatizar las simetrías de los orbitales fronterizos del metal y la olefina, dirigió a los químicos hacia una forma de simplificar las complejas interacciones entre los fragmentos, que ocurren dentro del marco orbital molecular, clasificando sus propiedades de simetría y agregando la importante restricción mecánica-cuántica de que solo interactúan orbitales con las mismas simetrías. Fukui reconoció la importancia de las interacciones orbitales fronterizas entre las moléculas orgánicas, y luego aplicó ampliamente los principios a una amplia gama de reacciones.
La propuesta de Dewar alentó a otros a reformular las formas canónicas de Pauling (Fig. 20) para los ligandos CO, NO, CN-, CNR, PF3, etc., dentro de un orbital molecular marco de referencia. El primer ejemplo de una representación pictórica de tipo Dewar para estos ligandos aparecieron en un artículo que describe la unión en [Ni(C5H5)(NO)] en 1956 por Orgel[79], y desde 1960, han sido ampliamente utilizados en libros de texto inorgánicos y organo-metálicos[80]. Orgel también fue influyente para complementar los argumentos de simetría de Dewar al proporcionar tablas de integrales de superposición para los orbitales 3d de metales de transición con los orbitales de valencia de ligandos comunes y otros metales[81]. Esto requirió la clasificación de las interacciones orbitales primarias σ, π y δ, que podrían usarse para calcular las superposiciones necesarias que incorporaron las relaciones angulares en moléculas específicas. Estos estudios establecieron claramente que la superposición entre los orbitales 3d y los orbitales 2p de B, C, N y NO fue significativa. Con el descubrimiento del ferroceno en 1952[82] y la determinación estructural, que estableció que tenía una estructura tipo sándwich, Orgel y Moffitt[83] de forma independiente pudieron utilizar los argumentos de simetría propuestos por Dewar y los datos de superposición para establecer un modelo orbital molecular para compuestos sándwich en general. Orgel también fue muy importante en la reformulación de los conceptos de campo de cristal desarrollados anteriormente para complejos de metales de transición en una teoría de campo de ligando basada en orbitales moleculares. Este análisis tuvo en cuenta las propiedades espectrales de los complejos de metales de transición y estableció la serie espectroquímica. Incluso en una etapa temprana, se reconoció que el CN- ??era alto en la serie espectroquímica y Orgel propuso que las propiedades ambifílicas de este y los ligandos relacionados con CO, CNR, NO contribuyeron a su mayor división orbital[84].
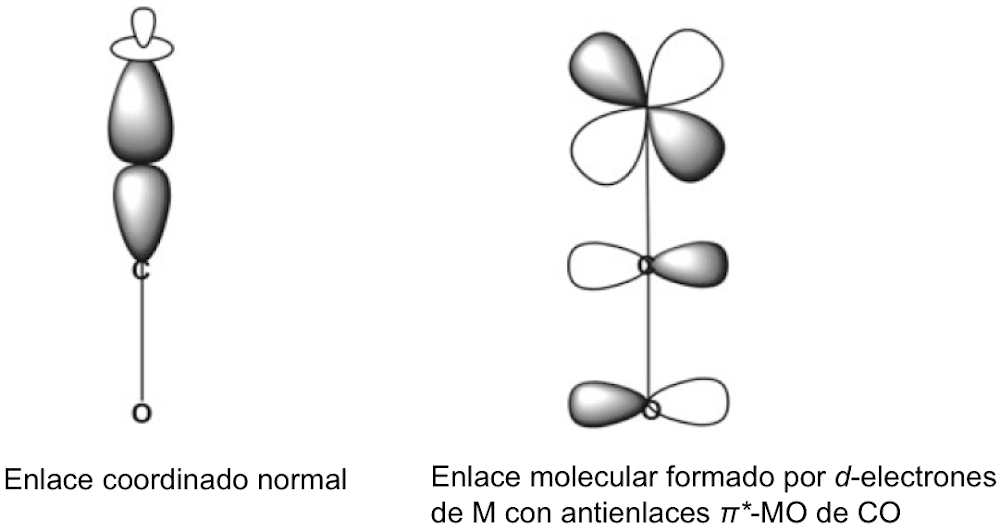
Fig. 20 Modelo de enlace sinérgico para ligandos aceptores π tales como CO, NO y CN-.
Se desarrollaron argumentos de simetría análoga cuando se estableció que los enlaces σ también podrían coordinarse con metales en estados de baja oxidación mediante el uso de orbitales análogos a los propuestos por Dewar para complejos de olefinas. Los complejos de dihidrógeno descubiertos por Kubas[85] fueron particularmente importantes para establecer este tipo de modo de coordinación, y las “interacciones agósticas” C-H que también se habían observado en varios complejos hipovalentes se racionalizaron utilizando este modelo. Las interacciones orbitales relevantes se ilustran en la Fig. 21 e implican la donación de los enlaces σ H2 o C–H y la donación inversa a los orbitales moleculares σ* ??antienlazamiento. Más recientemente, se ha demostrado que los complejos de metales de transición donde el metal tiene un estado de oxidación formal bajo son capaces de comportarse como bases de Lewis y formar complejos con ácidos de Lewis clásicos como AlCl3[86]. En estos complejos, el metal funciona como una base de Lewis utilizando un metal lleno dz^2 como se muestra en la figura 22. A partir de la década de 1970, el estudio de los complejos de metales de transición también estableció que ligandos como N, NR, O pueden complementar sus enlaces de coordenadas σ mediante donación π[87]. Durante el siglo pasado, el enlace de donante simple se ha convertido en una entidad mucho más compleja que implica la donación no solo de pares solitarios sino también de enlaces π y σ y la donación posterior de orbitales d metálicos a orbitales de antienlazamiento π* y σ* en el ligando e incluso de pares solitarios de metal a un ácido de Lewis. Las permutaciones de enlace que resultan de las interacciones de pares de electrones σ y π se resumen en la figura 23.
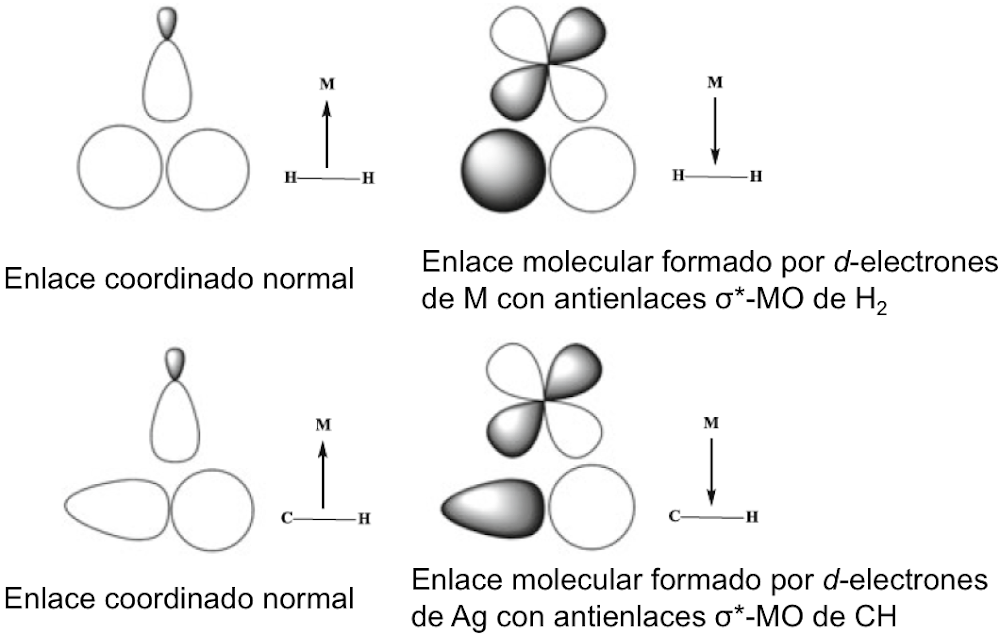
Fig. 21 Enlace sinérgico en complejos que implican enlaces σ, por ejemplo enlaces H2 y C–H.
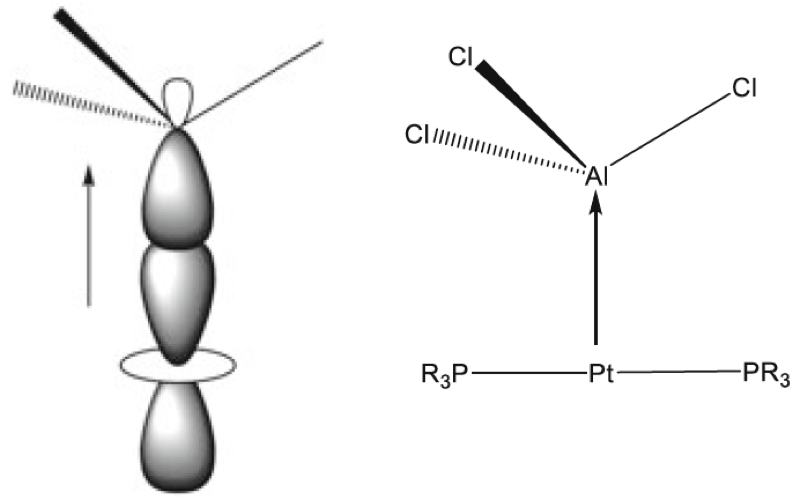
Fig. 22 Un ejemplo de un complejo que actúa como una base de Lewis hacia el clásico ácido de Lewis AlCl3.
1.10.4 Los cálculos de Ab Initio
Los documentos clásicos de Hartree, Fock, Roothan, Hall, Pople y Ahlrichs[88] sobre el método de campo autoconsistente permitieron por primera vez que los químicos soñaran con cálculos ab initio que lograron precisiones en la determinación de longitudes y ángulos de enlace y las propiedades termodinámicas de los enlaces que reproducen los datos experimentales con una precisión de 1 kcal/mol. Esto dio como resultado un grupo de químicos teóricos que estaban principalmente interesados ??en el cálculo a priori de las propiedades electrónicas de las moléculas pequeñas con este grado de precisión. Inicialmente, estos cálculos abordaron pequeñas moléculas de poco interés para los químicos experimentales promedio, pero el aumento exponencial del poder de las computadoras digitales siguió la ley de Moore, y dentro de una generación, fue posible lograr la precisión mencionada anteriormente para complejas moléculas orgánicas e inorgánicas.
La teoría moderna del enlace de valencia ahora complementa la teoría del orbital molecular, que no se adhiere a la idea del enlace de valencia de que los pares de electrones se localizan entre dos átomos específicos en una molécula, sino que se distribuyen en conjuntos de orbitales moleculares que pueden extenderse por toda la molécula. La teoría de la órbita molecular puede predecir las propiedades magnéticas y de ionización de manera directa, mientras que la teoría del enlace de valencia da resultados similares pero es más complicada. Los tratamientos de enlace de valencia están restringidos a moléculas relativamente pequeñas, en gran parte debido a la falta de ortogonalidad entre los orbitales de enlace de valencia y entre las estructuras de enlace de valencia, mientras que los orbitales moleculares son ortogonales. Por otro lado, la teoría del enlace de valencia proporciona una imagen mucho más precisa de la reorganización de la carga electrónica que tiene lugar cuando los enlaces se rompen y se forman durante el curso de una reacción química. En particular, la teoría del enlace de valencia predice correctamente la disociación de las moléculas diatómicas homonucleares en átomos separados, mientras que la teoría simple de la órbita molecular predice la disociación en una mezcla de átomos o iones. Por ejemplo, la función orbital molecular del dihidrógeno es una mezcla igual de las estructuras de enlace de valencia iónica y covalente, por lo que predice incorrectamente que la molécula se disociaría en una mezcla igual de átomos de hidrógeno e iones positivos y negativos de hidrógeno.
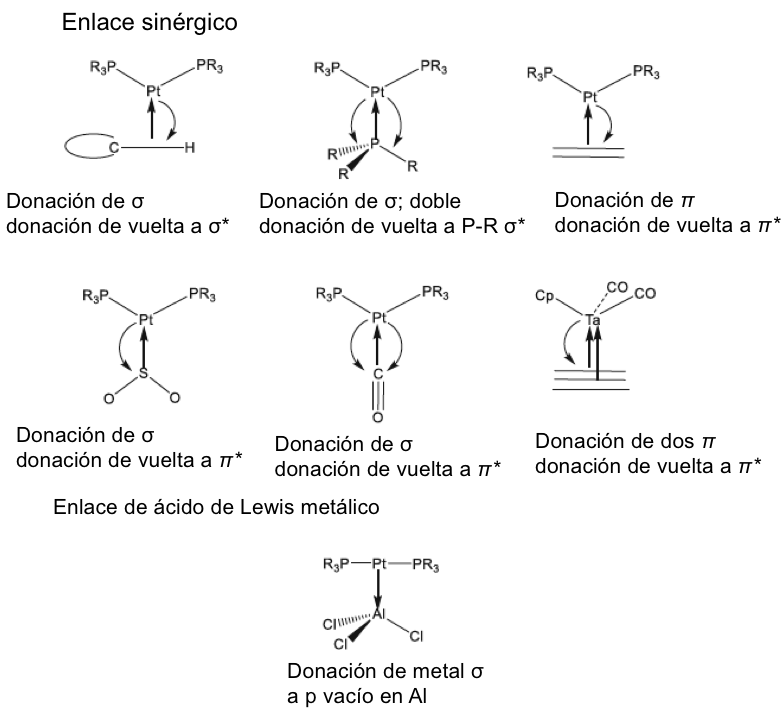
Fig. 23 Resumen de interacciones metal-ligando
La mayoría de los cálculos cuantitativos en la química cuántica moderna utilizan el enlace de valencia o la teoría molecular orbital como punto de partida, aunque un tercer enfoque, la teoría funcional de la densidad, se ha vuelto cada vez más popular en los últimos años. El impacto de la teoría funcional de la densidad en las últimas tres décadas ha sido sorprendente.
Su impacto en la química inorgánica ha sido particularmente dramático porque ha arrojado resultados confiables para moléculas complejas con orbitales de valencia d y f. Los cálculos contemporáneos de DFT combinan la descripción razonable de las estructuras electrónicas de compuestos de metales de transición con excelentes descripciones geométricas, y ha desplazado en gran medida el método “tradicional” de Hartree-Fock. Estos desarrollos han sido cubiertos en volúmenes anteriores de Estructura y Vinculación y el lector se refiere a estos desarrollos[89].
A principios de la década de 1970, surgió un nuevo enfoque de estructura electrónica desde la comunidad física y se describe como teoría funcional de densidad (DFT). La energía total de la molécula se expresó como funcional de la densidad electrónica total. Hohenburg y Kohn demostraron la relación única entre la densidad de electrones y la energía, y Kohn y Sham presentaron un enfoque práctico de DFT variacional. A pesar de que los cálculos sobre sólidos infinitos se habían informado desde la década de 1970, DFT no era considerado lo suficientemente preciso para los cálculos sobre moléculas hasta la década de 1990 cuando las aproximaciones utilizadas en la teoría se refinaron para describir exactamente el intercambio y correlación de interacciones. Costos computacionales para ab initio DFT los cálculos son relativamente bajos en comparación con el enlace de valencia y los métodos de órbitas molecular. DFT comenzó a acercarse a los objetivos de la termoquímica computacional para calcular las propiedades energéticas de los procesos químicos con una precisión de 1 kcal/mol. La aceptación generalizada de estos algoritmos por la comunidad química llevó a la concesión del Premio Nobel en 1998 a Kohn y Pople.
1.10.5 Enlaces orbitales naturales
El análisis de los resultados de cálculos orbitales moleculares deslocalizados para moléculas más complejas es problemático porque ya no existe una relación simple con las representaciones de enlaces localizadas por Lewis. El uso de análisis de fragmentos y poblaciones superpuestas ha sido útil, pero requiere cierto conocimiento de la teoría de perturbaciones. Se han desarrollado varios métodos para intentar cerrar la brecha entre los cálculos de los orbitales moleculares y las estructuras de Lewis, uno que se usa ampliamente son los orbitales de enlace natural (NBO). Cada enlace NBO σAB (el donante) se puede escribir en términos de dos híbridos de valencia dirigida (NHO) hA, hB en los átomos A y B, con los correspondientes coeficientes de polarización cA, cB:
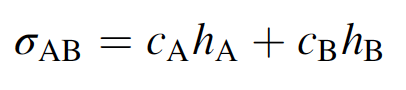
Los enlaces varían suavemente desde el límite covalente (cA = cB) hasta el iónico (cA >> cB). Cada NBO de enlace de valencia σ debe estar emparejado con un NBO de enlace de valencia correspondiente σ* (el aceptador) para completar el lapso del espacio de valencia:
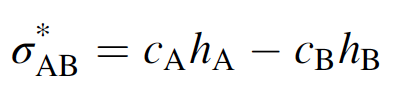
Las NBO de enlace del tipo “orbital de Lewis” tienen números de ocupación cercanos a 2, y las NBO antienlace son del tipo “no-orbital de Lewis” (números de ocupación cerca de 0).
En una estructura de Lewis idealizada, los orbitales de Lewis completos (dos electrones) se complementan con no-orbitales Lewis formalmente vacíos.
Se han desarrollado programas informáticos que pueden transformar los resultados de los cálculos moleculares orbitales en NBO. Una estructura de Lewis óptima se puede definir como aquella con la cantidad máxima de carga electrónica en los orbitales de Lewis (carga de Lewis). Una baja cantidad de carga electrónica en los orbitales de Lewis indica fuertes efectos de la deslocalización de electrones[90]. En las estructuras de resonancia, pueden existir estructuras contribuyentes mayores y menores. Estos análisis proporcionan resultados que son similares a los métodos modernos de teoría de enlace de valencia.
1.11 Resumen general
Los documentos de Lewis y Kossel y su posterior formulación en la matemática y el lenguaje de la mecánica cuántica tuvieron un gran impacto en la química. Su pensamiento ha proporcionado un marco conceptual, una notación y un lenguaje para discutir y evolucionar la química moderna. La redacción de algoritmos que incorporan la física que subyace a la mecánica cuántica y el desarrollo de computadoras modernas ha llevado a la posibilidad de calcular desde los primeros principios las propiedades de interés para los químicos (geometrías, propiedades espectroscópicas y sus perfiles de reacción) con precisión. Las generalizaciones de Lewis y Kossel surgieron en ausencia de un modelo de mecánica cuántica, pero no obstante han demostrado ser duraderas. Reunieron un buen conocimiento empírico de la literatura química y la capacidad de construir modelos conceptuales que explicaran las observaciones experimentales utilizando conceptos bien fundados en la mecánica cuántica moderna. Una característica particularmente importante ha sido la representación de estas ideas de una manera pictórica que los químicos encuentran fácil de apreciar y aplicar. Esto también requiere una notación y un medio de representación que permita a los químicos intercambiar ideas de manera creativa. Como McWeenie ha comentado, “si nuestros conceptos y “patrones de comprensión” son sólidos, serán respaldados por un cálculo riguroso; si no, deben ser rechazados[91]”. Llamó la atención sobre el siguiente párrafo del libro de Coulson[92]:
La química es un tema experimental cuyos resultados pueden incorporarse a un patrón en torno a conceptos bastante elementales. El papel de la química cuántica es comprender estos conceptos y mostrar cuáles son las características esenciales del comportamiento químico. Decir que la computadora electrónica muestra que D(H-F) >> D(F-F) no es una explicación en absoluto, sino simplemente una confirmación del experimento. Cualquier “explicación” aceptable debe ser en términos de repulsiones entre electrones que no se unen, fuerzas de dispersión entre “núcleos atómicos” y similares.
Los que han intentado responder a las preguntas fundamentales sobre la definición exacta de un enlace químico, los que han traducido la física básica en algoritmos eficientes y amigables con la computadora, y los que han desarrollado modelos conceptuales y los han presentado, han realizado progresos de manera complementaria de una manera que podría ser utilizada y adaptada por químicos experimentales. Los capítulos posteriores de esta serie de volúmenes de Estructura y Enlazamiento proporcionan revisiones actualizadas de los principales profesionales en el campo que amplifican estos objetivos y, con suerte, rinden homenaje adecuadamente a las principales contribuciones de Lewis y Kossel hace 100 años. Espero que sus ideas fomenten un progreso similar en el próximo siglo.
1.12 Configuración electrónica de elementos químicos
La configuración electrónica nos indica la distribución de los electrones en cada orbital de un elemento químico. Para poder realizar estas configuraciones es necesario conocer el diagrama de Moeller, el cual nos indicará qué orden seguirán los electrones al irse posicionando en cada uno de los orbitales del elemento químico de nuestro interés.

Los orbitales mostrados en el diagrama anterior son s, p, d, y f, sin embargo, existen más orbitales no presentes en este diagrama, pero para alcanzar esos orbitales se requiere de una cantidad muy grande de electrones, lo cual es una cualidad muy poco común de los elementos presentes en la tabla periódica.
La configuración electrónica va de la mano junto con los números cuánticos, pues estos podrán ser determinados si tenemos una configuración electrónica completa; a continuación, vamos a analizar un ejemplo:
Si escogemos al oxígeno (O), el primer paso es ver con cuántos electrones contamos (suponiendo un estado natural y neutralidad del átomo), entonces tendremos un número de protones = número de electrones, esto se representa con la letra z, por lo tanto para O tendremos z=8, ahora debemos tener en cuenta la capacidad de cada orbital para almacenar electrones, la cual es la siguiente:
Los exponentes en la configuración electrónica simbolizan la cantidad de electrones presentes en cada orbital. Por lo tanto, sumando exponentes y siguiendo el diagrama de Moeller vamos a tener la siguiente configuración electrónica:
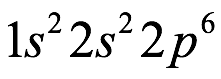
Pero los exponentes suman 2+2+6=10 y nosotros necesitamos que sumen 8, esto quiere decir que la última capa no se encuentra totalmente llena y en realidad la configuración real será:
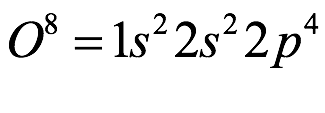
Entonces, como primer regla básica de la configuración electrónica: solo podemos cambiar el número de electrones presentes en el último orbital. Esta es válida como configuración electrónica, sin embargo, no es la más desarrollada y no nos permite obtener los 4 números cuánticos pues nos faltan datos, motivo por el cual vamos a representar de otra forma la configuración electrónica del oxígeno. En las configuraciones electrónicas los electrones se acomodan en pares sobre cada orbital, en el caso de que el orbital soporte más de un par de electrones, como es común, se separará ese mismo orbital en las partes que se requieran.
Por ejemplo, un orbital
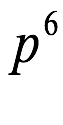
tiene capacidad para 3 pares de electrones, entonces se separan en
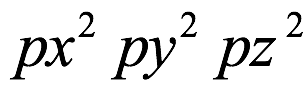
Donde xyz hacen referencia a los ejes geométricos en que se encuentran los electrones, no es necesario escribir xyz, simplemente se usa para diferenciar las posiciones de los electrones en el orbital p, cuando se va adquiriendo experiencia pueden ser omitidas esas literales. Los pares de electrones se representan con flechas hacia arriba y hacia abajo, siempre empezando con la que apunta hacia arriba, por ejemplo, en un orbital s los electrones se representan:
esto se debe al principio de exclusión de Pauli que en pocas palabras nos dice que un electrón no puede tener los mismos números cuánticos que otro, esto se explicará más adelante. Cuando se trata de un orbital p siempre se empieza rellenando con flechas hacia arriba
debido a la regla de Hund, la cual indica que los electrones se distribuyen siempre que sea posible lo más separados que puedan y una vez que se agote esta posibilidad empezará a rellenarse con flechas hacia abajo. Entonces regresando al ejemplo del oxígeno:
1er paso:
Representar los orbitales necesarios para la configuración en su forma separada por pares de electrones y vacía
2do paso:
Empezar a rellenando los orbitales que podemos completar
3er paso:
Rellenar con flechas individuales hacia arriba el último orbital
4to paso:
Rellenar con los electrones faltantes los espacios para flechas hacia abajo.
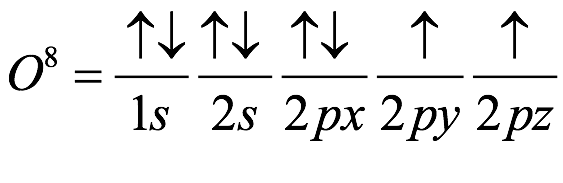
La configuración electrónica del oxígeno se encuentra ahora completa y de esta forma ahora somos capaces de determinar los números cuánticos del ultimo electrón.
Números cuánticos
En química los números cuánticos son 4; indican y se clasifican de la siguiente manera:
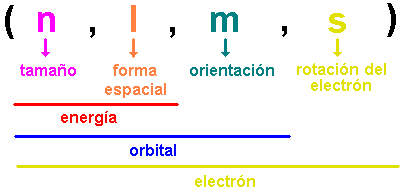
n: numero cuántico principal
Nos indica el nivel energético principal donde está ubicado el electrón y también una idea del volumen del orbital pues a mayor nivel mayor volumen. Por ejemplo, para el último electrón del oxígeno; n está dado por:
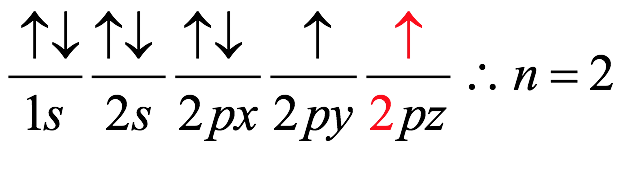
l: numero cuántico secundario
Nos indica la energía del subnivel (orbital: s,p,d,f…) en la que se encuentra el electrón donde
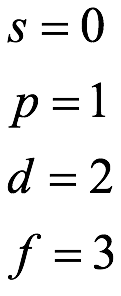
Por ejemplo, para el último electrón del oxígeno; l está dado por
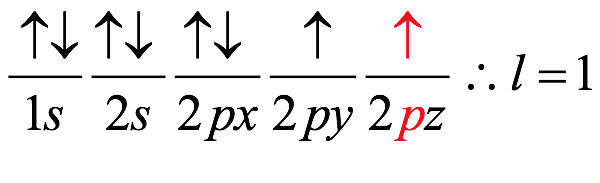
m: número cuántico magnético
Representa la probable orientación de los subniveles en el campo magnético del átomo
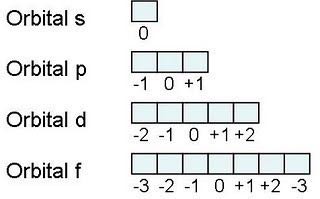
Entonces dependiendo de la posición del electrón dentro del orbital donde se encuentre se le asignara el número cuántico magnético. Por ejemplo, para el último electrón del oxígeno; m está dado por:


s: número cuántico de spin
Nos indica el sentido de giro del electrón con respecto al átomo; toma valores de,
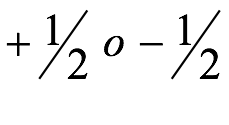 ,
,
si la flecha indica arriba entonces será positivo, por el contrario, será negativo. Por ejemplo, para el último electrón del oxígeno; s está dado por

aquí se puede ver claramente el por qué no puede haber dos flechas en el mismo par que apunten a la misma dirección, pues esto implicaría que todos sus números cuánticos son iguales y si se cambia la dirección de la flecha el electrón comparte todos los números cuánticos excepto el de spin, que tendría un cambio de signo y de esta forma se cumple el principio de exclusión de Pauli.
Con estos números cuánticos es posible determinar los elementos, pues el último electrón del último orbital de un elemento tiene números cuánticos diferentes a cualquier último electrón del último orbital de otro elemento.
Gases nobles
Los gases nobles tienen propiedades muy similares entre sí, por ejemplo a temperatura ambiente son gases, incoloros e inodoros y presentan una reactividad muy baja, de ahí surge su nombre, esto se debe a que todos excepto el helio presentan el octeto de electrones en su última capa de valencia y ya no requieren más electrones para ser estables. Los siete gases nobles son helio (He), neón (Ne), argón (Ar), kriptón (Kr), xenón (Xe), el radiactivo radón (Rn) y el sintético oganesón (Og).
Son muy útiles para la configuración electrónica pues todos ellos exceptuando al helio terminan su configuración en un subnivel p:
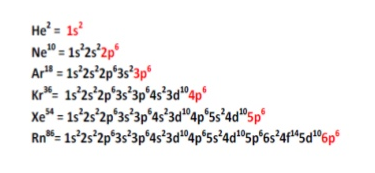
Entonces cuando se trata de configurar elementos con grandes cantidades de electrones se puede recurrir a las notaciones de Kernell, esto consiste en usar el gas noble con número de electrones inmediato inferior al elemento que se va a representar de la siguiente forma: tomando como ejemplo al potasio (K)
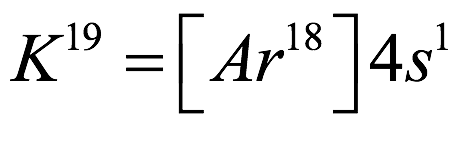
de esta forma ahorramos mucho tiempo y tinta en las configuraciones electrónicas.
Ejercicio:
Determine qué elemento químico tiene como números cuánticos en su último electrón:
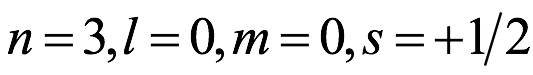
Solución:
1er paso
Con ayuda de los números n y l sabemos que el último electrón está en 3s y gracias al spin en este caso m siempre es 0 para los orbitales s y gracias al spin sabemos que la flecha apunta hacia arriba, entonces tendremos que nuestro electrón es
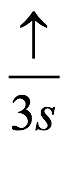
2do paso:
Consultar el diagrama de Moeller y ver dónde está 3s

Por la posición en la que está quiere decir que este elemento tiene lleno
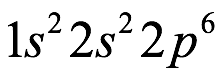 .
.
Entonces agregando el ultimo electrón tenemos que la configuración electrónica será
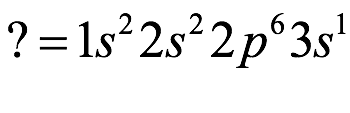
o en notación de Kernell
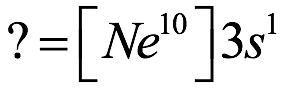
3er paso:
Finalmente determinaremos z que recordando significa el número de electrones presentes en el átomo, entonces 10+1=11 y buscamos en la tabla periódica que elemento tiene como número atómico 11; este resulta ser el sodio (Na)
Y solo queda anotar el elemento junto con su configuración electrónica
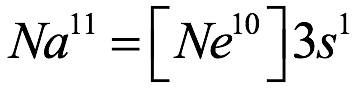
Ejercicio 1:
Determine qué elemento químico tiene como números cuánticos en su último electrón:
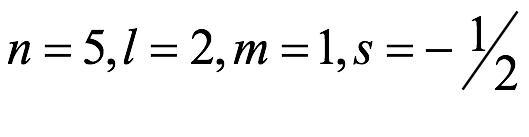
Solución:
1er paso
Con ayuda de n y l sabemos que el último orbital del que hablamos será 5d y como m=1 y el spin es negativo, podemos decir que este último orbital tendrá la distribución siguiente:

Nota: el último electrón aparentemente tiene spin positivo, pero se debe tener en cuenta la regla de Hund, por lo que realmente el último electrón será el marcado en rojo.
2do paso
Consultar el diagrama de Moeller y ver dónde está 5d

Por su posición podemos saber que
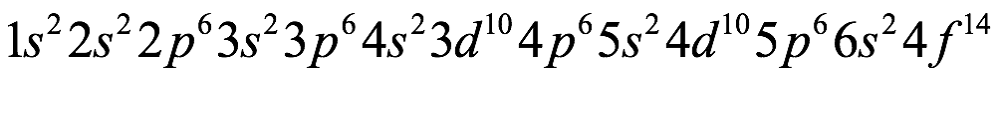
se encuentran llenos, por lo tanto, la configuración electrónica completa será:
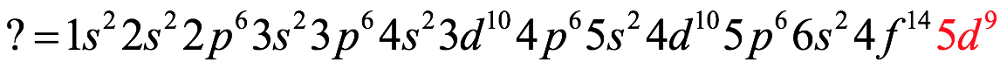
Observar que el último orbital no cuenta con los electrones suficientes para llenar el orbital, además, como se puede observar es una configuración electrónica bastante extensa, motivo por el cual es conveniente usar la notación de Kernell:
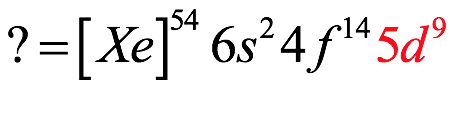
Si revisamos las notas el gas noble que más nos conviene usar es el xenón ya que al usar cualquier otro tendríamos que ampliar la notación o se rebasaría la cantidad de electrones que necesitamos.
3er paso
Finalmente determinaremos z que recordando significa el número de electrones presentes en el átomo, realizando la suma tendremos que: 54+2+14+9=79 y buscamos en la tabla periódica qué elemento tiene como número atómico 79; este resulta ser el oro (Au); Y solo queda anotar el elemento junto con su configuración electrónica
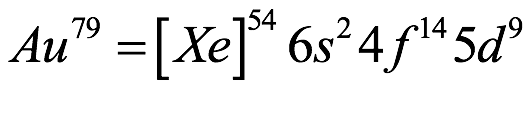
Autoevaluación:
Determine qué elemento químico tiene como números cuánticos en su último electrón:
a) 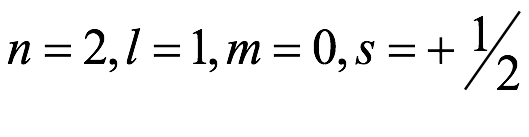 R=Carbono
R=Carbono
b) 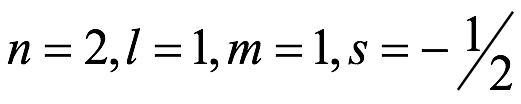 R=Nitrógeno
R=Nitrógeno
Obtenga los números cuánticos:
- a) Platino (Pt) R=
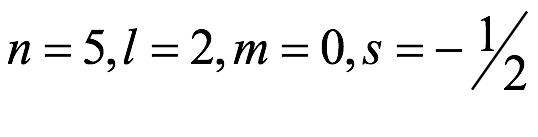
- b) Estaño (Sn) R=
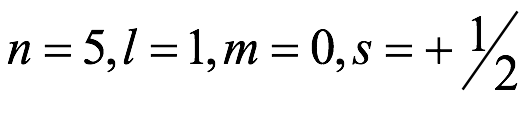
Referencias
[1] Knight, David. (2012). John Dalton (1766–1844). Philosophy of Chemistry. 71-77. 10.1016/B978-0-444-51675-6.50006-2.
[2] Trimble, Virginia. (2020). The Origin and Evolution of the Chemical Elements. 10.1142/9789811206467_0003.
[3] Zhu, Wansen & Fang, Caiyun & Xia, Lin. (2020). Chemical Elements in Life. 10.1142/11552.
[4] Jefferson, D.A. & Tilley, E.E.M.. (2020). The structural and physical chemistry of nanoparticles. 10.1201/9781003076896-5.
[5] Hoenig, Steven. (2019). Organic Chemistry. 10.1201/9780429277948-3.
[6] Chowdhury, Budhadeb & Malik, Susanta & Saha, Bidyut. (2020). Section Inorganic Chemistry. Journal of the Indonesian Chemical Society. 3. 131-138. 10.34311/jics.2020.03.3.131.
[7] Kenkel, John. (2020). Introduction to Analytical Chemistry. 10.1201/9781003069966-1.
[8] Palmiere, Cristian. (2020). Biochemistry. 10.1201/9780429188947-16
[9] Badash, Lawrence. (2004). Ernest Rutherford and the Explosion of Atoms. Isis. 95. 131-132. 10.1086/423554.
[10] Faura, Ferran & Iranipour, Shayan & Nocera, Emanuele & Rojo, Juan & Ubiali, Maria. (2020). The strangest proton?. The European Physical Journal C. 80. 10.1140/epjc/s10052-020-08749-3.
[11] Zhang, Tao. (2020). Relationship between band gap and equivalent volume of electron cloud. Modern Physics Letters B. 34. 2050400. 10.1142/S021798492050400X.
[12] Lin, Shiquan & Xu, Cheng & Liang, Xu & Wang, Zhong. (2020). The Overlapped Electron-Cloud Model for Electron Transfer in Contact Electrification. Advanced Functional Materials. 30. 10.1002/adfm.201909724.
[13] Mingos, D.Michael. (2016). The Chemical Bond: Lewis and Kossel’s Landmark Contribution. 10.1007/430_2015_203.
[14] Pulkkinen, Karoliina. (2019). Values in Action: Simplicity, Completeness, and Carefulness in the Development of the Systematisations of Chemical Elements. 10.17863/CAM.45920.
[15] Boeck, Gisela. (2019). Das Periodensystem der Elemente und Lothar Meyer. Chemie in unserer Zeit. 53. 10.1002/ciuz.201900001.
[16] Resnati, Giuseppe & Metrangolo, Pierangelo. (2020). Celebrating 150 years from Mendeleev: The Periodic Table of Chemical Interactions. Coordination Chemistry Reviews. 420. 213409. 10.1016/j.ccr.2020.213409.
[17] Davies, Alwyn. (2012). Sir William Ramsay and the Noble Gases. Science progress. 95. 23-49. 10.3184/003685012X13307058213813.
[18] Godun, R & Webb, C & Featonby, P & d'Arcy, M & Oberthaler, M & Summy, G & Foot, C & Burnett, Keith. (1999). Limits of the separated-path Ramsey atom interferometer. Journal of Physics B: Atomic, Molecular and Optical Physics. 32. 5033. 10.1088/0953-4075/32/20/317.
[19] Methven, S.. (2018). Ramsey, ‘Universals’ and atomic propositions. British Journal for the History of Philosophy. 27. 1-21. 10.1080/09608788.2018.1477731.
[20] Mingos, D.Michael. (2016). The Chemical Bond: Lewis and Kossel’s Landmark Contribution. 10.1007/430_2015_203.
[21] Snobelen, Stephen. (2007). The light of nature?: God and Natural Philosophy in Isaac Newton?s Opticks. 15-53.
[22] Park, Buhm Soon. (2017). SCientific Discoveries: Berzelius, Stadtman, And The Chemistry Of Selenium. Free Radical Biology and Medicine. 112. 4. 10.1016/j.freeradbiomed.2017.10.351.
[23] Rocke, A.. (1981). Kekulé, Butlerov, and the Historiography of the Theory of Chemical Structure. The British Journal for the History of Science. 14. 27 - 57. 10.1017/S0007087400018276.
[24] Dasgupta, Soumit & Mandalà, Marco & Guneri, Enis. (2020). Alexander Crum Brown: A Forgotten Pioneer in Vestibular Sciences. Otolaryngology Head and Neck Surgery. 163. 10.1177/0194599820937671.
[25] Hofmann, S. & Dmitriev, Sergey & Fahlander, C. & Gates, Jacklyn & Roberto, James & Sakai, Hideyuki. (2020). On the discovery of new elements (IUPAC/IUPAP Report): Report of the 2017 Joint Working Group of IUPAC and IUPAP. Pure and Applied Chemistry. -1. 10.1515/pac-2020-2926.
[26] Jensen, William. (2018). Richard Abegg and the Periodic Table. 10.1093/oso/9780190668532.003.0016.
[27] Bohr N (1922) Atomic structure and the chemical and physical properties of the elements. Z
Phys 9:1–67
[28] Sommerfeld A (1916) The Drude dispersion theory from the standpoint of Bohr’s model, and
the constitution of hydrogen, oxygen, and nitrogen. Ann Phys 51:1
[29] Kossel W (1916) Formation of molecules and its dependence on atomic structure. Ann Phys
49:229–362
[30] Langmuir I (1921) Types of valence. Science 54:59–67
[31] Pauli W (1927) Ueber Garsentartung der Paramagnetismum. Z Phys 31:765–785
[32] Lennard-Jones JK (1952) The spatial correlation of electrons in molecules. J Chem Phys
20:1024–1030
[33] Ingold CK (1922) The structure of the benzene nucleus. Part 1. Intra-nuclear tautomerism. J
Chem Soc Trans 121:1133–1143
[34] Robinson Sir Robert (1976) The memoirs of a minor prophet – 70 years of organic chemistry.
Elsevier, Oxford
[35] Bury CR (1921) Langmuir’s theory of the arrangement of electrons in molecules. J Am Chem
Soc 43:1602–1609
[36] Blanchard AA, Gulliland WL (1926) The constitution of nickel carbonyl and the nature of
secondary valency. J Am Chem Soc 48:872–882
[37] Sidgwick NV (1923) The nature of non-polar link. Trans Faraday Soc 19:469–475
[38] Reiff F (1931) Konstitution und Eigenschaften der Co(NO)(CO)3. Z Anorg Chem
202:375–381
[39] Jensen WB (2005) The origin of the 18 electron rule. J Chem Educ 82:28–29
[40] Green JC, Green MLH, Parkin G (2012) The occurrence and representation of three-centre
two-electron bonds in covalent inorganic compounds. J Chem Soc Chem Commun
48:11481–11503
[41] Sidgwick NV, Powell HW (1940) Stereochemical types valency groups. Proc R Soc London
Ser A 176:153–180
[42] Gillespie RJ, Robinson EA (2006) Gilbert N Lewis and the chemical bond: the electron pair
and the octet rule from 1916 to the present day. J Comput Chem 28:87–97
[43] Mingos DMP, Hawes JC (1985) Complementary spherical electron density model. Struct
Bond 63:1–63
[44] Lin Z (2016) Lewis description of bonding in transition metal complexes. Struct Bond.
doi:10.1007/430_2015_182
[45] Hoffman RV (2004) Organic chemistry an intermediate text. Wiley, New York
[46] Ghosh A, Berg S (2014) Arrow pushing in inorganic chemistry: a logical approach to the
chemistry of main group elements. Wiley, New York
[47] Longuet-Higgins HC, Bell RP (1943) The structure of boron hydrides. J Chem Soc 250–255
[48] Bartlett N (1962) Xenonhexafluoroplatinate. Proc Chem Soc, pp 218
[49] Lipscomb WN (1959) Recent studies on boron hydrides. Adv Inorg Radiochem 1:118–157
[50] Moeller T (1952) Inorganic chemistry. Wiley, New York
[51] Gillespie RJ, Nyholm RS (1958) Stereochemistry of inorganic molecules and complex ions
inorganic stereochemistry. Progr Stereochem (Academic, New York) 2:261–305
[52] Walsh AD (1953) The electronic orbitals, shapes and spectra of polyatomic molecules Part
I. J Chem Soc 2260–2265
[53] Hückel E (1931) Quantum theoretical contributions to the benzene problem I, II. The electron
configuration of benzene and related compounds. Z Phys 70(204–309):310–349
[54] Dewar MJS (1969) The molecular orbital theory of organic chemistry. McGraw-Hill,
New York
[55] Longuet-Higgins HC, de V Roberts M (1955) The electronic structure of an icosahedron of
boron atoms. Proc Roy Soc 230:110–119
[56] Elian M, Chen MML, Mingos DMP, Hoffmann R (1976) A comparative study of conical
fragments. Inorg Chem 15:1148–1155
[57] Weinhold F, Landis C (2005) Valency and bonding. Cambridge University Press, Cambridge,
pp 96–100
[58] Gavroglu K, Simoes A (1994) Early ideas in the history of quantum chemistry. Hist Stud Phys
Sci 25:47–110
[59] Burrau Ø (1927) Berechnung des Energiewertes des Wasserstoffmolekel-Ions (H2
+) im Normalzustand. Danske Vidensk. Selskab. Math.-fys. Meddel (in German) M 7:14: 1–18
[60] Heitler W, London F (1927) Interaction of neutral atoms according and homoplar binding
according to the quantum mechanics. Z Phys 44:455–472
[61] Pauli W (1922) U¨ ber das Modell des Wasserstoffmoleku¨lions. Ann Phys 373:177–240
[62] Hund F (1926) Zur Deutung einiger Erscheinungen in den Molekelspektren (On the interpretation
of some phenomena in molecular spectra). Z Phys 36:657–674
[63] James HM, Coolidge AS (1933) The ground state of the hydrogen molecule. J Chem Phys
1:825–835
[64] Pauling L (1928) The application of the quantum mechanics to the structure of the hydrogen
molecule and hydrogen molecule-ion and to related problems. Chem Rev 5:173–213
[65] Haaland A, TilsetM(2016) Lewis and Kossel’s legacy – structure and bonding in main group
compounds. Struct Bond. doi:10.1007/430_2015_192
[66] Li J, McWeeny R (2002) Pushing valence bond to new limits. Int J Quantum Chem
89:208–216
[67] Coulson CA, Fisher I (1949) Notes on the molecular orbital theory of the hydrogen molecule.
The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science: Series
7 40:386–393
[68] Goodgame MM, Goddard WA (1985) Modified generalized valence-bond method: a simple
correction for the electron correlation missing in generalized valence-bond wave functions;
prediction of double-well states for Cr2 and Mo2. Phys Rev Lett 54:661–664
[69] Herzberg G (1967) Molecular structure and molecular spectra. Van Nostrand, New York
[70] Fehlner TP, Bowser JR (1988) Proton power: an intuitive approach to the electronic structures
of molecule hydrides. J Chem Educ 65:976–980
[71] Woodward RB, Hoffmann R (1965) Stereochemistry of electrocyclic reactions. J Am Chem
Soc 87:395–397
[72] Dewar MJS (1969) The molecular orbital theory of organic chemistry. McGraw-Hill & Co,
New York
[73] Hoffmann R (1963) Extended Hu¨ckel theory I – hydrocarbons. J Chem Phys 39:1397–1412
[74] Pearson RG (1976) The symmetry rules for chemical reactions. Wiley, New York
[75] Hoffmann R (1988) Solids and surfaces: a chemist’s view of bonding in extended structures.
VCH, Weinheim
[76] HieberW(1942) The present status of the chemistry of metal-carbonyls. Die Chem 55:24–28
[77] Chatt J, Willians AA (1951) The nature of the co-ordinate link IV. Complex formation by
phosphorus trifluoride. J Chem Soc 3061–3078
[78] Wilkinson G (1951) The preparation and properties of tetrakistribromophosphine-nickel and
tetrakistrifluorophosphinenickel. J Am Chem Soc 73:5501–5502
[79] Orgel LE (1956) Electronic structures of some mixed compounds of cyclopentadienyl and
carbon monoxide or nitric oxide with the transition metals. J Inorg Nucl Chem 2:315–322
[80] on FA, Wilkinson G (1962) Advanced inorganic chemistry: a comprehensive text.
Intersciene/Wiley, New York
[81] Craig DP, Macoll A, Nyholm RS, Orgel LE (1954) Chemical bonds involving d orbitals I. J
Chem Soc 332–353
[82] Wilkinson G, Rosenblum M, Whiting MC, Woodward RB (1952) The structure of iron bis
(cyclopentadienyl). J Am Chem Soc 74:2125–2156
[83] Moffitt W (1954) The electronic structure of bis-cyclopentadienyl compounds. J Am Chem
Soc 76:3386–3392
[84] Orgel LE (1955) Spectra of transition metal complexes. J Chem Phys 23:1004–1014
[85] Kubas G (2014) Activation of dihydrogen and co-ordiantion of molecular H2 to transition
metals. J Organomet Chem 751:33–49
[86] Braunschweig H, Gruss K, Radachki K (2007) Interaction between d and p block metals.
Synthesis and structure of platinum alane adducts. Angew Chem Int Ed 46:7782–7784
[87] Winkler JR, Gray HB (2012) Electronic structures of oxo-metal ions. Struct Bond 142:17–28
[88] Hinchcliffe A (2000) Modelling molecular structures, 2nd edn. Wiley, Chichester
[89] Kaltsoyanis N, McGrady JE (2004) Principles and applications of density functional theory in
inorganic chemistry I. Struct Bond 112:1–189
[90] Weinhold F, Landis CR (2012) Discovering chemistry with natural bond orbitals. Wiley,
New Jersey
[91] McWeeny R (1979) Coulson’s valence, 3rd edn. Oxford University Press, Oxford
[92] Coulson CA (1961) Valence, 2nd edn. Oxford University Press, Oxford
Autores:
Eduardo Ochoa Hernández
Nicolás Zamudio Hernández
Abraham Zamudio Durán
Lizbeth Guadalupe Villalon Magallan
Mónica Rico Reyes
Pedro Gallegos Facio
Gerardo Sánchez Fernández
Rogelio Ochoa Barragán